
Abstract
Activated microglia plays an important role in monitoring the microenvironment and prune neural process in healthy neural tissue, in order to maintain synaptic homeostasis. However, hyperactive microglia may release various cytotoxic factors and induce neuroinflammation, which cause neuronal damages leading to neurodegenerative diseases. Tanshinone IIA (TSA), an extract from traditional Chinese medicine, features potent anti-apoptotic and anti-inflammatory effects both in vitro and in vivo. But little is known on the effects of TSA on microglial-over-activation-induced neural impairments. In this study, by employing murine BV2 cell lines as well as the combinations of ELISA assay, immunostaining, western blotting analysis and RT-PCR, we found that TSA has the potential to exhibit anti-inflammatory effects. We hereby demonstrated that TSA rescued neural growth and development in the primary cultured hippocampal neurons from impairments caused by BV2 microglial over-activation insult. The results show that TSA attenuated the BV2 cell activation by lipopolysaccharide (LPS) stimulation through suppressing the NF-кB signal pathway. Also, conditioned mediums (CM) from TSA treated and activated BV2 cells protected against LPS-CM-induced neuronal death. Furthermore, TSA treatment could recover the inhibitory effects of LPS-CM on growth cone extension, neurite sprouting and outgrowth, as well as spinogenesis. Our findings support that TSA is capable of inhibiting BV2 cell over-activation thus has potential protective effects in the cultured hippocampal neurons. This study may lay a foundation for using TSA to restore cerebral injuries after severe neuroinflammation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Danshen (DS) is a traditional Chinese medicine derived from dried root of Salvia miltiorrhiza Bunge. Extensive studies have revealed that DS features potent anti-apoptotic and anti-inflammatory effects [1]. Tanshinone IIA (TSA), one of the most effective lipophilic diterpene extracts of DS, exhibits a variety of biochemical activities including protection against ischemia–reperfusion (I/R) injury, oxidative stress-triggered damage and stabilization of vascular endothelial functions [2, 3]. Its antioxidant properties have been demonstrated to repair lipid peroxidation both in vitro and in vivo [4, 5]. TSA also exerts neuroprotective effects in hypoxia–ischemia-damaged neonatal brain [6]. Besides the protection from brain injuries, TSA inhibits the proliferation, induces apoptosis and exhibits potent cytotoxicity against tumors such as NCI-H460 cells [7] and leukemia cells [8]. These beneficial effects of TSA strongly support its therapeutic potential in the near future.
Microglial cells are of the monocytic lineage and comprise approximately 12 % of total cells found in the brain, and function similarly to the macrophages of the systemic immune system. Microglia typically exists in a quiescent state in the normal brain, characterized by ramified morphology and short branched processes [9, 10]. However in a severely disrupted microenvironment, microglia become over-activated, transforming into a migratory amoeboid shape and leading to extensive phagocytosis [11]. Consequently, a variety of cytotoxic factors including arachidonic acid derivatives like prostaglandin E2 (PGE2) and pro-inflammatory cytokines are released, promoting the initiation or escalation of neurodegeneration [12, 13]. The expression of cyclooxygenase-2 (COX-2) is predominately responsible for the production of PGE2. In cultured microglia from the mouse brain, lipopolysaccharide (LPS) was found to induce microglial over-activation and contributes to the expression of COX-2 [13]. This process can be blocked by inhibitors of nuclear factor-kappa B (NF-κB), a ubiquitous transcription factor best known for its role in regulating immune and cell-survival/death pathways. NF-κB activation in neurons is mediated primarily by the binding of p50 and p65 to the κB DNA recognition sequence. Interleukin-1β (IL-1β) and pro-inflammatory cytokines tumor necrosis factor-α (TNF-α) are known as stimuli that more strongly activate neuronal NF-κB, than other agents like L-glutamate [14, 15]. Therefore, inhibitors of the NF-κB pathway may become important alternatives in preventing further anti-neuroinflammation in the microglial over-activation.
Currently there hasn’t been any study on the capability of TSA to rescue the neural impairments induced by microglial over-activation. Therefore in this study, we aimed to investigate the beneficial effects and the underlying mechanisms of TSA treatment on the impairments of neural growth and development, in primary cultured hippocampal neurons using LPS-induced BV2 microglial over-activation as an in vitro model.
Methods and Materials
Cell Culture
For the microglial cell culture, the murine BV2 cells at a density of 1 × 105/ml were maintained in Dulbecco’s Modified Eagle Medium (DMEM; Gibco, USA) supplemented with 10 % fetal bovine serum (FBS; Gibco, USA), 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco) at 37 °C in a humidified incubator under 5 % CO2. For LPS stimulation, cells were cultured 24 h and then treated with LPS (100 ng/ml) for another 24 h.
For the primary cultured neurons, the hippocampus of both hemispheres was dissected from the brain of postnatal days 1 ICR mouse, removed and collected in Falcon tubes in 0.25 % trypsin. The tissue was digested in trypsin for 15 min at 37 °C, and then gently triturated mechanically by using a sterile pipette with a wide opening to dissociate larger aggregates. The cells were seeded at high density (20,000 cells/cm2) for quantification of survival at 7 days in vitro and at low density (2000 cells/cm2) for measurement of neurite lengths and spine density in vitro on poly-l-lysine-coated Petri dishes or in 96/24-well culture plates (Corning, USA). The cells were plated in DMEM-F12 medium containing 10 % fetal calf serum, 2 % B-27 supplement, penicillin–streptomycin, Ham’s F-12 Nutrient Mixture. Cultures were maintained at 37 °C in humidified atmosphere with 5 % CO2 and half of the cell culture medium was replaced every 4 days. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee on the Ethics of Animal Experiments of The Second Xiangya Hospital. The IACUC committee members at The Second Xiangya Hospital approved this study. All surgery was performed under sodium pentobarbital anesthesia, and all efforts were made to minimize suffering.
Conditioned Medium (CM) Collection
For CM collection, BV2 cells were cultured in microglia medium for 24 h, cell culture supplement replaced by neuronal medium with or without TSA (Xian Guanyu Bio-tech, China) and/or LPS (Sigma, 100 ng/ml) stimulation. TSA was dissolved in phosphate-buffered saline (PBS) with 1 % DMSO at a stock solution of 20 mM and stored at −20 °C until use. The final concentration of DMSO in all experiments is less than 0.001 %. For all the control cultures, normal microglia medium was added instead of TSA. CM was harvested from free-floating cells by centrifugation at 1000×g for 5 min. After this step, CM was sterile filtered and stored at −80 °C until use.
MTT Assay and LDH Assay
The MTT assay was used to detect cells viability under different TSA concentration, while the membrane integrity of neurons was measured by Lactase Dehydrogenase (LDH) assay. The standard protocol is followed by the CytoTox-ONE Homogeneous Membrane Integrity Assay kit. After 7 days of culture, the assays were carried and data were collected by using Multi label Reader (Victor 4, PerkinElmer, Singapore).
Enzyme-Linked Immunosorbent Assay (ELISA)
ELISA kit (RD System, USA) was used to determine the concentrations of TNF-α and IL-1β in culture supernatants. The standard procedure follows the manufacturer’s instruction. Then, the microplate reader was used to measure the absorbance at 450 nm. Therefore, the concentrations of TNF-α and IL-1β in culture supernatants calculated based on the standards of TNF-α and IL-1β.
Nitric Oxide (NO) Production
NO concentrations in cell culture supernatants were determined by measuring nitrite, which is a major stable product of NO, using Griess reagent (Beyotime, China). Cells were stimulated with or without LPS for 24 h, then, 50 μl culture medium was mixed with an equal volume of Griess reagent. The absorbance at 540 nm was determined using a microplate reader. A standard nitrite curve was generated in the same fashion using NaNO2 standards.
RT-PCR
After incubation, media was removed and BV2 cells were washed with phosphate-buffered saline (PBS, pH 7.4) twice. Total RNA was extracted using Trizol reagent as recommended by the manufacturer (Invitrogen, USA). RNA quality and concentration were evaluated spectroscopically using a NanoDrop 2000c instrument (Thermo Scientific, USA). For mRNA analysis, real-time PCR was performed using Superscript ™-ш kit (Invitrogen) on an ABI 7900HT PCR machine, and data were normalized to β-actin and further normalized to the control without LPS stimulation unless otherwise indicated. The sequences of PCR primers were as follows:
-
1.
iNOS: sense 5′-GGACGAGACGGATAGGCAGAGATT-3′, antisense 5′-AAGCCACTGACACTTCGCACAA-3′;
-
2.
COX-2: sense 5AGCCACTGACACTTCGCACAA-3′′, antisense 5′-GCACGTAGTCTTCGATCACT-3′;
-
3.
β-actin: sense 5′-GATGGTGGGAATGGGTCAGA-3′, antisense 5′-TCCATGTCGTCCCAGTTGGT-3′.
Data analysis was performed using the 2−ΔΔCt method.
Immunostaining
The cells were treated following the protocols of the Fast ImmunoFluorescence Staining Kit (BPIF30-1KT, Protein Biotechnologies, USA). At least six plates of cells were immunostained for each group. The primary antibodies used were as follows: anti-actin (Sigma) and anti-β-tubulin (Sigma).
Western Blotting
Cells were washed with ice-cold PBS twice, then scraped, collected and transferred into clean centrifuge tubes. Nuclei and cytosol were separated using a nuclear extract kit according to the manufacturer’s instructions (Active Motif). The expressions of p65 (MW 65 kD), IкBα (MW 35 kD) and p-IкBα (MW 35 kD) involved in NFкB signaling pathway in cytoplasm and nucleus were measured by western blotting and β-actin (MW 42 kD) was used as control. HDAC1 (MW 55 kD) was employed as the control of p65 expression in nucleus. Cell extracts were lysed in RIPA buffer (Beyotime) containing Complete Protease Inhibitor Cocktail and 2 mM PMSF. About 20 μg of total protein was separated by 10 % SDS-PAGE and then transferred to 0.45 μm Nitrocellulose Membrane (Millipore). Protein concentrations were determined by BCA Protein Assay Kit (Beyotime).The membranes were incubated with primary antibodies at 4 °C overnight and then hybridized with appropriate HRP-conjugated secondary antibody (Amersham Pharmacia Biotech) at room temperature for 2 h. Protein signals were visualized using ECL detection system. The primary antibodies used were as follows: anti-p65, anti-HDAC1, anti-β-actin (Santa Cruz Biotechnology), anti-GAP43 (Sigma), anti-IκBα and anti-p-IkBα (Epitomics).
Transfection and Morphology Analysis
Fluorescence images were acquired either by confocal laser scanning microcopy (Zeiss LSM710) or Fluorescence Imaging Device (Olympus-FSX100). The length of the dendrites and filopodia were determined using ImageJ. At least 80 neurons per group were counted in each experiment and three independent experiments were performed. To visualize the growth cones, neurons were fixed for 10 min with 2 % PFA and stained with Alexa Fluor 488-conjugated phalloidin probe (Invitrogen) and neuron-specific β-tubulin (anti-Tuj1) (R&D systems). Data were collected from three independent experiments. For the transfection, the cultured hippocampal neurons were transfected with mCherry-actin at DIV17 by the calcium phosphate method and the images were taken at DIV21 by confocal laser scanning microscopy.
Statistical Analysis
All the experiments were repeated at least three times independently, and data were presented as the mean ± SEM based on the independent experiments. The two-way analysis of variance (ANOVA) test was used for statistical analysis, and then followed by Tukey test (LST) to estimate the statistical difference between different groups. The probability values of *p < 0.05 and # p < 0.01 were considered as significance.
Results
Effects of TSA on BV2 Cell Viability
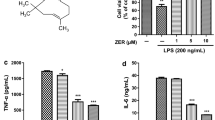
Prior to studying the anti-inflammatory effects of TSA, we needed to find the optimal dose of TSA treatment for the BV2 cell viability in vitro. It was previously reported that BV2 cells in the substrates featured diverse morphological properties [16]. Figure 1a shows the typical morphology of BV2 cells in this study. MTT and LDH assays were performed after 1 day of incubation in different concentrations of TSA (1, 5, 10 and 20 μM). We found that 10 and 20 μM TSA significantly decreased the cell viability, while 1 and 5 μM TSA failed to induce any obvious cell death (Fig. 1b). Moreover the cell viability in TSA-treated groups exhibited a dose-dependent manner. This was further validated by LDH results. LDH release is an assay signal to indicate the state of cell death [17]. Higher LDH release represents more aggravated cell injury. As expected, we found that the release of LDH is gradually elevated by the increased TSA concentrations (Fig. 1c). TSA concentrations of up to 5 μM had no significant effect on the LDH release. However, the LDH release was strongly enhanced at higher doses (10 and 20 μM, Fig. 1c). These results indicated TSA treatment at higher concentrations (>5 μM) may induce cell death and the concentration employed should not exceed this value. Therefore to avoid the toxic effect of TSA on cell growth, 5 µM TSA was selected for the following investigations as it imposed negligible effects on cell growth.

Effects of TSA exposure at different doses on BV2 cell viability. a Representative photograph of BV2 cells in the culture without TSA treatment. Scale bar 200 µm. b Relative cell viability of the BV2 cells to the control (without LPS and TSA treatment) following different doses of TSA treatment (0, 1, 5, 10, 20 µM), measured by MTT assay. c Relative LDH release to control in the experimental groups. The experiments were repeated in triplicate. The data were presented by the mean ± SEM. *p < 0.05
Effects of TSA Treatment on the LPS-Induced Inflammatory Responses
To explore the possible pro-inflammatory response during TSA treatment, we characterized the levels of TNF-α, IL-1β and NO released into the culture medium. TNF-α is best characterized by its role in mediation of systemic inflammation, stimulating the acute phase reaction [18]. IL-1β is also an important mediator of the inflammatory response and is involved in a variety of cellular activities, including cell proliferation, differentiation and apoptosis [19]. Moreover it has been shown to exert beneficial effects on the proliferation, survival, differentiation and migration of neural precursor cells [20]. Figure 2a, b clearly illustrated that the levels of TNF-α and IL-1β remained invariable under non-LPS treatment. After LPS stimulation (100 ng/ml) for 24 h, the amount of TNF-α and IL-1β in the culture were strongly boosted. In the presence of LPS, we found that 5 µM TSA co-treatment significantly reduced the release of TNF-α and IL-1β in cells, clearly suggesting the ability of TSA in rescuing LPS-induced inflammation. Moreover, the expression of inducible nitric oxide synthase (iNOS) and COX-2, both of which are important enzymes involved in the regulation of microglia inflammation [16], were examined by RT-PCR. iNOS plays a critical role in inflammation since it controls the production of NO [21]. COX-2 can be massively detected at the site of inflammation as a key enzyme involved in the release of prostaglandins regulating inflammation [22, 23]. The expression of iNOS and COX-2 were clearly elevated under LPS induction when compared with the non-LPS-treated group. It is noticeable that 5 µM TSA treatment significantly reduced the expressions of iNOS and COX-2 in LPS group (Fig. 2c, d), consistent with the earlier TNF-α and IL-1β results in Fig. 2a, b. Similar results could also be observed in the NO contents in the experimental groups. LPS treatment caused much higher NO contents in the media compared to the control, while 5 µM TSA significantly attenuated the LPS-induced increase of NO contents (Fig. 2e).

Effects of TSA treatment (5 µM) on the LPS-induced inflammatory responses in the BV2 cells. The contents of cytokines, including TNF-α (a) and IL-1β (b) in the experimental groups were measured by the ELISA assay. Note the obviously lower concentrations of TNF-α and IL-1β in TSA treatment group compared to untreated group after LPS stimulation. c, d Relative mRNA levels of iNOS and COX-2 in the experimental groups assessed by real time PCR using the 2−ΔΔCT method. e NO contents in BV2 cells in different groups, measured by means of Griess reaction. Data are expressed as the fold change in gene expression normalized to an endogenous gene (β-actin) and relative to the cells in control group without LPS stimulation. All the experiments were repeated three times. Data are presented as mean ± SEM (n = 7). # p < 0.01
TSA Inhibited NF-кB Signaling Pathway
To further explore the underlying mechanisms of the inhibition of inflammatory responses in BV2 cells by TSA treatment, we examined the activation of NF-кB signaling pathway during LPS stimulation with or without TSA treatment.
First, the expressions of p65, IкBα and p-IкBα proteins in the cytoplasm of cells from the experimental groups was analyzed by western blotting. As shown in Fig. 3a, the expression of p65 and IкBα proteins in the BV2 cells was decreased after LPS stimulation, while 5 µM TSA co-treatment returned their expressions to levels comparable to that of the control. Accordingly, the expression of p-IкBα in the cytoplasm was evidently enhanced after LPS stimulation and the 5 µM TSA treatment reduced it to a similar extent as control. The results provided consistent findings about the expressions of these three kinds of proteins normalized to cytoplasmic marker β-actin during LPS induction and TSA treatment (Fig. 3b–d). Furthermore, p65 levels in the nucleus was enhanced by LPS stimulation, as normalized to nuclear marker HDAC1, indicating the occurrence of p65 translocation into the nucleus and the activation of NF-κB in LPS-stimulated BV2 cells, while TSA treatment restored the elevated p65 level in the nucleus (Fig. 3e, f). These results validated the inhibitory effects of TSA treatment on LPS-induced BV2 cell activation, suggesting that TSA may inhibit the NF-кB signaling pathway involved in the inflammation responses.

TSA (5 µM) inhibited NF-кB signaling pathway activated by LPS stimulation. a The p65, IкBα and p-IкBα protein expression in the cytoplasm in the cultures under TSA treatment with or without LPS stimulation. The relative optical densities of p65 (b), IкBα (c) and p-IкBα (d) protein bands to control (without LPS and TSA treatment), normalized to β-actin. e The p65 expression in the nucleus in the experimental groups. f The relative optical densities of p65 protein bands to control, normalized to HDAC1. The experiments were repeated in triplicate. Data were presented by mean ± SEM. *p < 0.05, # p < 0.01
TSA Improved Neuronal Survival After LPS-CM Culturing
It was reported that the over-activated microglia has abnormal functions and promotes neurodegeneration [18], we therefore studied the possible protective effects of TSA on the neuronal death induced by BV2 cell over-activation. The CM was collected and employed for the following experiments (see the Materials and Methods section), since the CM contains secreted chemokines and cytokines by activated microglia [16]. CM from BV2 cell enriched cultures in the absence or presence of 5 µM TSA were prepared in the presence and absence of LPS stimulation. Cell viability of the cultured hippocampal neurons in different CM for 7 days was examined by MTT and LDH assay. The cell viability in the LPS-CM group was significantly reduced compared to the control group (Fig. 4a), indicating LPS-induced BV2 cell over-activation may be detrimental to the neurons. In contrast, 5 µM TSA co-treatment greatly improved cell viability in the LPS-CM. Results from LDH release assay were also consistent with the above findings (Fig. 4b). Significantly higher level of LDH was released in LPS-CM cultured neurons in comparison with the control group, whereas TSA treatment reversed this elevated LDH release, suggesting that the cell viability was improved after TSA treatment.

TSA (10 µg/ml) rescued LPS-CM-induced cell death in the hippocampal neurons. The cells were cultured for 7 days in the different conditioned mediums of the experimental groups. a Relative cell viabilities to control in the experimental groups, examined by MTT assay. b Relative LDH release to control (without LPS and TSA treatment) in the experimental groups. The experiments were repeated in triplicate. Data were presented by mean ± SEM. *p < 0.05
TSA Rescued the Impairments of GCs by LPS-CM Treatment
Another aim of this study is to investigate the effects of TSA treatment on the impairments of neuronal growth by BV2 cell over-activation. We explored the growth cone (GC) formation and extension of the neurons cultured in different CM for 24 h. At this time point, the neurons do not form a complex neural network, therefore provide a suitable time window to observe the GCs. The morphology of GCs was observed under fluorescence microscope by actin and β-tubulin labeling. Figure 5a shows representative images of GCs with actin-rich peripheral zone (green) and microtubule-rich central core (red). The normalized area of GCs was clearly reduced by LPS-CM treatment, while this effect was inhibited by the addition of TSA in LPS-CM (Fig. 5b). The number of filopodia of each GC was also evidently decreased in LPS-CM group by approximately 40 %, and it could be rescued by TSA treatment as indicated by a significant increase in the number of filopodia (Fig. 5c). However, TSA may repair this effect by promoting the formation of filopodia, accounting for the increase of the number of filopodia in each GC by TSA treatment. Although no change of filopodial length was observed between different CM-treated groups (Fig. 5d), the density of filopodia (number of filopodia/GC area) was reduced in LPS-CM cultured neurons compared to the control group (Fig. 5e). GC density was significantly increased by TSA treatment in LPS-CM cultured neurons.

TSA (5 µM) rescued the impairments of GCs by LPS-CM stimulation. The hippocampal neurons were cultured for 1 day in the different conditioned mediums of the experimental groups. a Representative fluorescence images of the hippocampal GCs in the cultures of experimental groups stained for actin, tubulin and merge of the two staining. Scale bar 20 µm. b Relative area of GCs to control (without LPS and TSA treatment). c Average number of filopodia emerging from GCs. d Average filopodium lengths from the tip of each filopodia to the edge of the GCs. e Ratio of number of filopodia and area of GC in GCs. The experiments were repeated in triplicate. Data were presented by mean ± SEM. *p < 0.05
Effects of TSA on Neurite Sprouting and Outgrowth
The neurons began to extend neurites after being seeded on the substrates when they were mature [24]. Figure 6a shows typical hippocampal neurons with extending neurites on the substrate after 7 days of culturing, stained for microtubule-associated protein 2 (MAP-2) (marker for mature neurons). Neurites, as the main structure of a neuron, refer to either a dendrite or an axon under different conditions, and reflect the healthy status of cell growth both in vitro and in vivo. In order to study the effects of TSA treatment on the neurite sprouting and outgrowth, three main indexes were measured and analyzed in the experimental groups: the number of primary dendrites per cell, dendritic end tips and the average neurite length. A significant decrease in the primary dendrites per neuron was observed in the LPS-CM cultured cells compared to the control, while 5 µM TSA co-treatment rescued this neuritic loss (Fig. 6b). Similarly, LPS-CM culturing clearly reduced the numbers of dendritic end tips and the average length of neurites compared to the control, indicating an inhibitory effects on neuritic outgrowth (Fig. 6c, d). As expected, TSA treatment exhibited a protective role against the LPS-CM-induced impairments by promoting the number of dendritic tips, as well as the averaged neuritic length, to normal control levels (Fig. 6c, d). These results provided strong evidences that TSA protects neurite sprouting and outgrowth in the event of BV2 cell over-activation. Furthermore, the expression of GAP43 in the cultures of different groups was analyzed by western blotting. GAP43 exists in nervous tissue-specific cytoplasm and is considered to serve as an important protein in neurite formation and regeneration [25]. The expression of GAP43 in the culture was significantly reduced in LPS-CM group compared to the control (Fig. 6e, f). Of note, TSA treatment led to a significant increase of GAP43 expression compared to LPS-CM group (Fig. 6e, f). This observation likely accounted for the promoted neuritic outgrowth and sprouting after being damaged by LPS-CM, however the potential molecular mechanisms of increment of GAP43 expression remain to be studied.

TSA treatment (5 µM) could repair the LPS-CM-induced injuries of neurite sprouting and outgrowth. The hippocampal neurons were cultured for 7 day in the different conditioned mediums of the experimental groups. a Typical hippocampal neurons with extending neurites in the culture of experimental groups, stained for MAP-2. The neurite sprouting and outgrowth were characterized by the number of primary dendrites per cell (b), number of dendritic end tips (c) and the average neurite length (d) in the experimental groups. e Western blotting analysis of GAP-43 expression in the cultures of different groups. f Relative optical densities of GAP-43 show in (e) (n = 3/group). The experiments were repeated in triplicate. Data were presented by mean ± SEM. *p < 0.05, # p < 0.01
Effects of TSA on Spinogenesis
Finally, we investigated the effects of TSA treatment on spinogenesis by observing the morphological changes of cells and their density in the cultures of different CM. The neurons were cultured for 17 days and were transfected with mCherry-actin. At day 21, the neurons with spines can be clearly visualized by confocal microscopy. Figure 7a shows the images of the neurite with spines under a higher magnification in all of the four experimental groups. The process of spine maturation starts from a filopodium-like protrusion to a mushroom shape with a post-synaptic density located at the tip. Dendritic spines, which can be categorized into three morphological categories (stubby, mushroom and thin), are considered as the primary loci of synaptic plasticity, identified as small extensions shorter than 3 µm from the adjacent dendrite. The proportions of the three spine types (stubby, mushroom and thin) were compared among the experimental groups, and no significant difference was observed (Fig. 7b), indicating that neither LPS-CM nor TSA treatment could affect the spine morphology in mature neurons. However, the spine density in the culture in LPS-CM was obviously lower compared to that of control (Fig. 7c). Remarkably increased spine density was also found in the hippocampal neurons after 5 µM TSA treatment. These results supported the protective role of TSA against the impairments of inflammation on neural development.

Effects of TSA treatment (5 µM) on spine morphology and density in the hippocampal neurons cultured for 14 days. a The cultured hippocampal neurons were transfected with mCherry-actin and imaged. The spine could be clearly observed at high magnification in these four experimental groups. b Proportion of different spine types expressed as percentage of total spines in the neurons in the experimental groups. c Spine density expressed as number of spines per 10 µm of dendrites of the neurons in the experimental groups. The experiments were repeated in triplicate. Data were presented by mean ± SEM. *p < 0.05
Discussion
In this study, murine BV2 cells were used as an in vitro model for primary microglia culture. These cells, which are able to secret lysozyme, interleukin 1 and tumor necrosis factor, retain most of the phenotypical and morphological properties featured by isolated microglial cells, and are best characterized by their phagocytic ability, esterase and lacked peroxidase activity [26]. Our study demonstrated that TSA at higher concentration (>5 μM) may induce BV2 cell injury (Fig. 1), and is consistent with previous study that demonstrated TSA had cytotoxic effects in vivo in tumor growth to lung tissue of breast cancer cells and human umbilical vein endothelial cells when administered at 5, 10 and 50 μM [16].
Many studies have addressed the correlations between the microglial activation and pro-inflammatory cytokines release, such as TNF-α, IL-1β, IL-6 and NO, which are primarily provoked by activated microglia during inflammatory response and detected at the cell lesion site [18]. Here, LPS was used to induce BV2 cells activation, since microglial over-activation has been most effectively demonstrated in models using LPS [27]. LPS was employed in this study based on two reasons. On one hand LPS-induced BV2 cells over-activation serves as an ideal positive control for us to evaluate the anti-inflammatory effects of TSA. On the other hand, we can investigate the possible involvement and potential mechanism underlying TSA-altered neuron behaviors in the BV2 cell culture after LPS-induced over-activation.
In response to an infection or injury, microglia acquire a reactive inflammatory phenotype, also referred to as the classic phenotype. The classic inflammatory phenotype is characterized by increased microglia proliferation, morphological changes and the release of several inflammatory molecules including cytokines, chemokines, superoxide and NO [28]. Our results regarding the cytokines, enzymes and NO release clearly demonstrated the capability of TSA in inhibiting the inflammatory responses, in the in vitro model of LPS-induced BV2 cell over-activation (Fig. 2). In fact, the anti-inflammatory effects of TSA were also reported by other researchers [29, 30]. Fang et al. [31] revealed that TSA not only inhibited the oxidation process but also attenuated the inflammation in atherosclerotic lesion, by suppressing the MMP-2 activity and the expression of CD40.
The other major finding in our current study was the molecular mechanisms underlying the TSA-inhibited inflammatory response in BV2 cells. As an important transcription factor, NF-κB can be activated by the exposure to inflammatory cytokines or LPS simulation. The activated form of NF-κB is a heterodimer, usually consisting of two subunits p65 and p50 [32]. NF-κB is also thought to bind IкBα and IкBβ, which serve to block its nuclear translocation. Upon binding to its DNA target sequence, NF-κB activates the transcription of genes involved in inflammation and immune response, as well as genes that control cell growth. Since NF-κB activation is promoted by IкBα kinase (IKK)-NF-κB-inducing kinase (NIK) pathways and mitogen-activated protein kinases (MAPK) pathways [32], TSA is likely to inhibit the LPS-induced NF-κB activation by suppressing NIK, IKK and MAPK pathways, which includes p38, JNK and ERK1/2. Our results indicated that the NIK-IKK pathways are likely to be involved in the mechanisms underlying the inhibitory effects of TSA in the primary cultured hippocampal neurons. Cao et al. [33] have previously demonstrated that TSA alleviates spinal nerve ligation induced neuropathic pain through the MAPK pathways. In addition, TSA also exerts anti-inflammatory effects via estrogen receptor (ER) dependent pathways in LPS activated RAW 264.7 cells. It functions as an efficient ER modulator by suppressing the expression of iNOS genes and NO production [34]. Taken together these findings provide comprehensive insights into the underlying mechanism of the anti-inflammatory effects of TSA. Moreover, our current study further indicated that TSA exerted its anti-inflammatory roles through suppressing NF-κB signaling pathway (Fig. 3).
TSA was also reported to exhibit neuroprotective effects. Qian et al. [35] found that TSA protected against β-amyloid protein 42 toxicity in cortical neurons, where the CM based experimental approach was also employed in the investigation. Our results from neuronal death as well as neuronal growth and development clearly demonstrated that TSA-treated-CM rescued the impairments by BV2 cell over-activation (Figs. 4, 5, 6, 7). The protective effects of TSA against CM-induced neuronal death may be attributed to the reduced amount of cytokines released into the CM upon TSA treatment (Fig. 2), since accumulated cytokines were shown to be detrimental to cell viability [18]. Furthermore, some important parameters reflecting neuronal growth and development were characterized in the presence of TSA-treated-CM, including GC, filopodia, neurite and spine. GCs, formed by an extended lamellipodium from where thin filopodia emerge [36], are highly motile structures located at the tip of the neurites, translating multiple extracellular guidance cues into directional movements [37–39]. Filopodia, which radiate out from the central core of GC, function as sensors to detect the surrounding environment [40, 41]. The primary function of filopodia is to sense the factors released by other cells in the surrounding environment, and transduce the signals to their parent growth cone. Therefore disruption of filopodia growth is likely to cause deficits in axonal transport, resulting in various brain disorders such as chronic neurodegenerative disease [42]. Our results demonstrated BV2 cells over-activation impaired GC extension, filopodia density, neurite density, spine density and pattern, and TSA was able to protect neural development against LPS-CM-induced inflammation (Fig. 5, 6, 7).
In summary, we hereby demonstrate that TSA exhibits anti-inflammatory effects through inhibiting NF-кB signal pathways, and exerts protective roles against the BV2 cell over-activation-induced impairments on cell viability, GC extension, neuritic sprouting and outgrowth, and spinogenesis in primary culture of hippocampal neurons. Many neurodegenerative diseases or acute injuries in the nervous system result in loss of neurons and damage of neurites and spines. Based on our observations, TSA may be potentially used as alternative therapeutic approach to protect cells from this insult.
Abbreviations
- TSA:
-
Tanshinone IIA
- DS:
-
Danshen
- LPS:
-
Lipopolysaccharide
- CM:
-
Conditioned mediums
- GC:
-
Growth cone
- NO:
-
Nitric oxide
- I/R:
-
Ischemia–reperfusion
- PGE2 :
-
Prostaglandin E2
- COX-2:
-
Cyclooxygenase-2
- NF-Κb:
-
Nuclear factor-kappa B
- IL-1β:
-
Interleukin-1β
- TNF-α:
-
Tumor necrosis factor-α
References
Zhang X, He D, Xu L, Ling S (2012) Protective effect of tanshinone IIA on rat kidneys during hypothermic preservation. Mol Med Rep 5:405–409
Wu Y, Chen Y, Zhang Z, Dong Y, Yu X, Jiao J, Kang Y, Gao W, Lou J, Liu Y (2009) Study on ultrastructure of cardioprotection of ramipril against ischemia/reperfusion injury in diabetic rats. Chin J Appl Physiol 25:485–489
Fu J, Huang H, Liu J, Pi R, Chen J, Liu P (2007) Tanshinone IIA protects cardiac myocytes against oxidative stress-triggered damage and apoptosis. Eur J Pharmacol 568:213–221
Zhou G-Y, Zhao B-L, Hou J-W, Ma G-E, Xin W-J (1999) Protective effects of sodium tanshinone IIA sulphonate against adriamycin-induced lipid peroxidation in mice hearts in vivo and in vitro. Pharmacol Res 40:487–491
Niu X-L, Ichimori K, Yang X, Hirota Y, Hoshiai K, Li M, Nakazawa H (2000) Tanshinone II-A inhibits low density lipoprotein oxidation in vitro. Free Radic Res 33:305–312
Xia WJ, Yang M, Fok TF, Li K, Chan WY, Ng P-C, Ng HK, Chik KW, Wang CC, Gu GJS (2005) Partial neuroprotective effect of pretreatment with tanshinone IIA on neonatal hypoxia–ischemia brain damage. Pediatr Res 58:784–790
Hu H, Zhang Y, Huang F, Deng H (2005) Inhibition of proliferation and induction of apoptosis by tanshinone II A in NCI-H460 cell. J Chin Med Mater 28:301–304
Song Y, Yuan S, Yang Y, Wang X, Huang G (2005) Alteration of activities of telomerase in tanshinone IIA inducing apoptosis of the leukemia cells. China J Chin Mater Med 30:207–211
Fetler L, Amigorena S (2005) Neuroscience. Brain under surveillance: the microglia patrol. Science 309:392–393
Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308:1314–1318
Levine ES, Zam A, Zhang P, Pechko A, Wang X, FitzGerald P, Pugh EN, Zawadzki RJ, Burns ME (2014) Rapid light-induced activation of retinal microglia in mice lacking Arrestin-1. Vis Res 102:71–79
Hanisch UK, Kettenmann H (2007) Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci 10:1387–1394
Velagapudi R, Aderogba M, Olajide OA (2014) Tiliroside, a dietary glycosidic flavonoid, inhibits TRAF-6/NF-κB/p38-mediated neuroinflammation in activated BV2 microglia. Biochim Biophys Acta Gen Subj 1840:3311–3319
Listwak S, Rathore P, Herkenham M (2013) Minimal NF-κB activity in neurons. Neuroscience 250:282–299
Liu Y-W, Huang Y-T (2014) Inhibitory effect of tanshinone IIA on rat hepatic stellate cells. PLoS ONE 9:e103229
Song Q, Jiang Z, Li N, Liu P, Liu L, Tang M, Cheng G (2014) Anti-inflammatory effects of three-dimensional graphene foams cultured with microglial cells. Biomaterials 35:6930–6940
Lobner D (2000) Comparison of the LDH and MTT assays for quantifying cell death: validity for neuronal apoptosis? J Neurosci Methods 96:147–152
Luo X-G, Ding J-Q, Chen S-D (2010) Microglia in the aging brain: relevance to neurodegeneration. Mol Neurodegener 5:12
Wang X, Fu S, Wang Y, Yu P, Hu J, Gu W, Xu XM, Lu P (2007) Interleukin-1beta mediates proliferation and differentiation of multipotent neural precursor cells through the activation of SAPK/JNK pathway. Mol Cell Neurosci 36:343–354
Wang X, Fu S, Wang Y, Yu P, Hu J, Gu W, Xu X-M, Lu P (2007) Interleukin-1β mediates proliferation and differentiation of multipotent neural precursor cells through the activation of SAPK/JNK pathway. Mol Cell Neurosci 36:343–354
Aktan F (2004) iNOS-mediated nitric oxide production and its regulation. Life Sci 75:639–653
Seibert K, Zhang Y, Leahy K, Hauser S, Masferrer J, Perkins W, Lee L, Isakson P (1994) Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proc Natl Acad Sci USA 91:12013–12017
Crofford LJ (1997) COX-1 and COX-2 tissue expression: implications and predictions. J Rheumatol Suppl 49:15–19
Brandt N, Franke K, Rašin MR, Baumgart J, Vogt J, Khrulev S, Hassel B, Pohl EE, Šestan N, Nitsch R (2007) The neural EGF family member CALEB/NGC mediates dendritic tree and spine complexity. EMBO J 26:2371–2386
Li N, Zhang X, Song Q, Su R, Zhang Q, Kong T, Liu L, Jin G, Tang M, Cheng G (2011) The promotion of neurite sprouting and outgrowth of mouse hippocampal cells in culture by graphene substrates. Biomaterials 32:9374–9382
Blasi E, Barluzzi R, Bocchini V, Mazzolla R, Bistoni F (1990) Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J Neuroimmunol 27:229–237
Russo I, Barlati S, Bosetti F (2011) Effects of neuroinflammation on the regenerative capacity of brain stem cells. J Neurochem 116:947–956
Kettenmann H, Hanisch UK, Noda M, Verkhratsky A (2011) Physiology of microglia. Physiol Rev 91:461–553
Throckmorton DC, Brogden AP, Min B, Rasmussen H, Kashgarian M (1995) PDGF and TGF-bold beta mediate collagen production by mesangial cells exposed to advanced glycosylation end products. Kidney Int 48:111–117
Kim SK, Jung K-H, Lee B-C (2009) Protective effect of tanshinone IIA on the early stage of experimental diabetic nephropathy. Biol Pharm Bull 32:220–224
Z-y Fang, Lin R, B-x Yuan, Yang G-D, Liu Y, Zhang H (2008) Tanshinone IIA downregulates the CD40 expression and decreases MMP-2 activity on atherosclerosis induced by high fatty diet in rabbit. J Ethnopharmacol 115:217–222
Il Jang S, Jin Kim H, Kim Y-J, Jeong S-I, You Y-O (2006) Tanshinone IIA inhibits LPS-induced NF-κB activation in RAW 264.7 cells: possible involvement of the NIK–IKK, ERK1/2, p38 and JNK pathways. Eur J Pharmacol 542:1–7
Cao F-L, Xu M, Wang Y, Gong K-R, Zhang J-T (2015) Tanshinone IIA attenuates neuropathic pain via inhibiting glial activation and immune response. Pharmacol Biochem Behav 128:1–7
Fan G-W, Gao X-M, Wang H, Zhu Y, Zhang J, Hu L-M, Su Y-F, Kang L-Y, Zhang B-L (2009) The anti-inflammatory activities of tanshinone IIA, an active component of TCM, are mediated by estrogen receptor activation and inhibition of iNOS. J Steroid Biochem Mol Biol 113:275–280
Qian YH, Xiao Q, Xu J (2012) The protective effects of tanshinone IIA on beta-amyloid protein (1-42)-induced cytotoxicity via activation of the Bcl-xL pathway in neuron. Brain Res Bull 88:354–358
Mongiu AK, Weitzke EL, Chaga OY, Borisy GG (2007) Kinetic-structural analysis of neuronal growth cone veil motility. J Cell Sci 120:1113–1125
Bray D, Thomas C, Shaw G (1978) Growth cone formation in cultures of sensory neurons. Proc Natl Acad Sci USA 75:5226–5229
Goodman CS (1996) Mechanisms and molecules that control growth cone guidance. Annu Rev Neurosci 19:341–377
Song H, Poo M (2001) The cell biology of neuronal navigation. Nat Cell Biol 3:E81–E88
Davenport RW, Dou P, Rehder V, Kater SB (1993) A sensory role for neuronal growth cone filopodia. Nature 361:721–724
Kater SB, Rehder V (1995) The sensory-motor role of growth cone filopodia. Curr Opin Neurobiol 5:68–74
Chevalier-Larsen E, Holzbaur EL (2006) Axonal transport and neurodegenerative disease. Biochim Biophys Acta 1762:1094–1108
Conflict of interest
The authors declare that they have no competing interests.
Ethical standard
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Wang, Y., Yang, L. & Yang, D. Tanshinone IIA Rescued the Impairments of Primary Hippocampal Neurons Induced by BV2 Microglial Over-Activation. Neurochem Res 40, 1497–1508 (2015). https://doi.org/10.1007/s11064-015-1624-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-015-1624-z