
Abstract
Hypoglycemia can cause neuronal cell death similar to that of glutamate-induced cell death. In the present paper, we investigated the effect of glucose removal from incubation medium on changes of mitochondrial and plasma membrane potentials in rat brain synaptosomes using the fluorescent dyes DiSC3(5) and JC-1. We also monitored pH gradients in synaptic vesicles and their recycling by the fluorescent dye acridine orange. Glucose deprivation was found to cause an inhibition of K+-induced Ca2+-dependent exocytosis and a shift of mitochondrial and plasma membrane potentials to more positive values. The sensitivity of these parameters to the energy deficit caused by the removal of glucose showed the following order: mitochondrial membrane potential > plasma membrane potential > pH gradient in synaptic vesicles. The latter was almost unaffected by deprivation compared with the control. The pH-dependent dye acridine orange was used to investigate synaptic vesicle recycling. However, the compound’s fluorescence was shown to be enhanced also by the mixture of mitochondrial toxins rotenone (10 µM) and oligomycin (5 µg/mL). This means that acridine orange can presumably be partially distributed in the intermembrane space of mitochondria. Glucose removal from the incubation medium resulted in a 3.7-fold raise of acridine orange response to rotenone + oligomycin suggesting a dramatic increase in the mitochondrial pH gradient. Our results suggest that the biophysical characteristics of neuronal presynaptic endings do not favor excessive non-controlled neurotransmitter release in case of hypoglycemia. The inhibition of exocytosis and the increase of the mitochondrial pH gradient, while preserving the vesicular pH gradient, are proposed as compensatory mechanisms.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hypoglycemia is a common consequence of diabetes mellitus treatable by insulin [1–3]. About 25–30 % of patients suffer from hypoglycemic episodes according to certain assessments [3]. Non-compensated severe hypoglycemia promptly leads to coma, depression of electrical activity of the brain and death [2–4]. However, recurrent hypoglycemia can lead to adverse events as well. It causes cognitive performance impairment, mood disorders and selective neuronal cell death in different brain regions [3]. Characteristics of hypoglycemia-induced neuronal cell damage closely resemble those of excitotoxic glutamate-induced death [2, 3, 5]. Furthermore, hypoglycemia was reported to be averted by application of N-methyl-d-aspartate (NMDA)—receptor antagonists [6]. Glucose deprivation can be used as an in vitro model for hypoglycemia [7–9].
Withdrawal of energy substrates from incubation medium caused a fast decrease of mitochondria potential in cultured cortical neurons and isolated neuronal presynaptic endings termed synaptosomes [7, 8]. This treatment also produced the decrease of plasma membrane potential, although the effect was less pronounced [7, 8]. It is well known that the mitochondrial proton-motive force (ΔµH+) includes two components, membrane potential (ΔΨ) and pH gradient across the inner membrane of mitochondria (ΔpH) [10–12]. It was proposed that the decrease of ΔΨ during hypoglycemia can be compensated by the increase of ΔpH [7], however experimental evidence supporting this assumption is still lacking.
In addition to mitochondria, neuronal presynaptic terminals contain synaptic vesicles, another type of organelles having electrochemical and pH gradients across the vesicular membrane [13–15]. They have acid intravesicular pH, which are generated by V-ATPase. Providing that synaptic vesicles have considerably higher surface/volume ratio over mitochondria or presynaptic terminals [15], an energy deficit would be expected to cause a more rapid dissipation of ion gradients in this organelle. Dissipation of the pH gradient across the synaptic vesicle membrane inhibits neurotransmitter uptake and depletes their cytosolic pool. Moreover, this can induce neurotransmitter efflux via plasma membrane transporters [16–18]. This phenomenon can potentially underlie excitotoxic brain damage during hypoglycemia.
In the present paper, we tested the hypothesis that the main biophysical characteristics of neuronal presynaptic endings upon glucose deprivation favor excessive non-controlled neurotransmitter release followed by the death of neighboring neurons due to excitotoxicity.
Isolated neuronal presynaptic endings termed synaptosomes were used as the main object. They preserved many properties of intact terminals including synaptic vesicle recycling, calcium-dependent neurotransmitter release and have the same ion gradients [15, 19, 20].
The plasma membrane potential was monitored by the fluorescent dye DiSC3(5) [21], the mitochondrial potential was determined by the fluorescent dye JC-1 [22], synaptic vesicle recycling and pH gradients across internal membranes were measured by the fluorescent dye acridine orange [14].
Materials and Methods
Materials
Oligomycin, 3,3′-dipropylthiadicarbocyanine [DiSC3(5)], carbonyl cyanide 3-chlorophenylhydrazone (CCCP), antimycin A, bafilomycin A1 were purchased from Sigma (St. Louis, MO, USA). 4-(2-Hydroxyethyl)piperazine-N′-1-ethanesulfonic acid (HEPES) was obtained from Merck (Darmstadt, Germany). Acridine orange was obtained from Serva (Heidelberg, Germany). 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolo-carbocyanine iodide (JC-1) and rotenone were obtained from Calbiochem (La Jolla, CA, USA). Tris(hydroxymethyl)aminomethane (tris) was obtained from BDH (Poole, UK).
Synaptosomes Preparation
Synaptosomes were isolated from brain hemispheres of 12–16-week-old male Wistar rats according to Hajos [23]. Stock suspensions of synaptosomes (10 mg/mL) were prepared in medium A (composition in mM: 132 NaCl, 5 KCl, 10 glucose, 1.3 MgCl2, 1.2 NaH2PO4, 15 HEPES, 5 tris, pH 7.4, 310 mOsm/l) and kept on ice. Animal experiments were carried out in accordance with EU Directive 2010/63/EU.
Hypoglycemia Model
The incubation of synaptosomes in medium A without glucose for 30 min at 37 °C was used as the hypoglycemia model. Pellet was resusupended in glucose-free medium A for these experiments. Synaptosomes were transferred to ice after 30 min incubation and used within 2 h. Glucose-free medium was used throughout the fluorescence measurements.
Determination of Intrasynaptosomal Mitochondria Membrane Potential
The membrane potential of intrasynaptosomal mitochondria was detected by the fluorescent dye JC-1 according to Chinopoulos et al. [22] with modifications according to Pekun et al. [24].
Synaptosome purification was carried out in calcium-free medium A and the pellet was resuspended in the same medium (protein concentration of 5 mg/mL). The suspension was incubated for 30 min at 37 °C in the presence of 10 μg/mL dye. Extracellular dye was washed once by sedimentation and the final pellet was resuspended in 2.0 mL calcium-free medium A (protein concentration of 10 mg/mL).
To investigate mitochondrial membrane potential, 200 μL of loaded synaptosomes were added to a cuvette containing 1.8 mL of the incubation medium A with 2 mM CaCl2. Fluorescence intensity was recorded at λex/em = 504/535 nm using a spectrofluorimeter Cary Eclipse (“Varian”, USA) at constant stirring and 37 °C. Rotenone (10 µM) + oligomycin (5 µg/mL) were added to the cuvette at 50 s. The same quantity of vehicle was added during control experiments. The control curve was extracted from the experimental curve.
Investigation of Plasma Membrane Potential
The plasma membrane potential was investigated using the fluorescent dye 3,3′-dipropylthiadicarbocianyne (DiSC3(5)) according to Waseem and Fedorovich [21]. An aliquot of synaptosome suspension (200 μL) was added to the cuvette containing 2 mL of the incubation medium A with 2 mM CaCl2. After 1 min 1 μM of DiSC3(5) was added to the cuvette. Fluorescence intensity was recorded at λex/em = 640/688 nm again using a spectrofluorimeter Cary Eclipse (“Varian”, USA) at constant stirring and 37 °C. KCl was added to the cuvette 1 min after addition of the dye. The same quantity of NaCl was added during control experiments. The control curve was extracted from the experimental curve.
Investigation of pH Gradients Across Intrasynaptosomal Membranes and Characterization of the Synaptic Vesicle Cycle
pH gradients and exocytosis were detected using fluorescent dye acridine orange according to Zoccarato et al. [14] with some modifications [19].
Fluorescence was recorded using spectrofluorimeters Cary Eclipse (“Varian”, USA) at 37 °C in constant stirring. Recording parameters were as follows: λex—494 nm, λem—529 nm. 200 μL of synaptosomal suspension were added to 1800 μL medium A containing 2 mM CaCl2 to the final protein concentration of 1 mg/mL. Next, 20 μL of aqueous solution of acridine orange were added to the final concentration of 5 μM. Suspension was incubated for 10 min to allow equilibration between synaptosomes and dye. Thereafter, various additions were made. The same quantity of vehicle was added during control experiments. The control curve was extracted from the experimental curve.
Other Methods
Protein concentration was assayed according to Lowry et al. [25] using bovine serum albumin as a standard. Data are presented as mean ± SEM where indicated, statistical significance was evaluated using one-tailed Student’s t test.
Results
Influence of Glucose Withdrawal from Incubation Medium on Intrasynaptosomal Mitochondria Potential
Figure 1a, b show that mitochondrial toxins rotenone + oligomycine or antimycine + oligomycine are able to increase fluorescence of JC-1. Therefore this dye can be used for investigation of intrasynaptosomal mitochondria membrane potential.

Influence of glucose withdrawal on JC-1 fluorescence. a Kinetics of JC-1 fluorescence increase after rotenone (10 µM) + oligomycin (5 µg/mL) addition. Rotenone + oligomycin (R + O) were added, when indicated. Curves represent 14 independent experiments. Con—control, GW—synaptosomes were incubated 30 min at 37 °C in medium without glucose. b Kinetics of JC-1 fluorescence increase after antimycin A (2 µM) + oligomycin (5 µg/mL) addition. Antimycin + oligomycin (A + O) were added, when indicated. Curves represent eight independent experiments. Con—control, GW—synaptosomes were incubated 30 min at 37 °C in medium without glucose. c Influence of glucose withdrawal on JC-1 fluorescence. Con—control, R + O/GW—glucose withdrawal; intrasynaptosomal mitochondria were depolarized by rotenone + oligomycin; synaptosomes were incubated 30 min at 37 °C in medium without glucose. A + O/GW—glucose withdrawal; intrasynaptosomal mitochondria were depolarized by antimycin A + oligomycin; synaptosomes were incubated 30 min at 37 °C in medium without glucose. Bars represent JC-1 fluorescence increase within 4 min after addition of rotenone (10 µM) + oligomycin (5 µg/mL) or antimycin A (2 µM) + oligomycin (5 µg/mL). Data presented are mean values ± SEM of at least 8 experiments. 100 % level corresponds to fluorescence increase in glucose-containing control medium. **P ≤ 0.01 versus 100 %
Figure 1a–c show that incubation of synaptosomes without glucose during the 30 min is leading to the decrease of the fluorescent dye JC-1 uptake by mitochondria at 40 %. The dye was released by rotenone + oligomycine or antimycine + oligomycine induced depolarization.
Glucose removal reduces mitochondria depolarization by toxins. It is suggested that glucose deprivation depolarizes intrasynaptosomal mitochondria.
Influence of Glucose Withdrawal from Incubation Medium on Synaptosomal Plasma Membrane Potential
In our previous work [21, 26, 27], 60 mM potassium chloride has been used to study rapid changes in synaptosomal plasma membrane potential. This concentration could well be insufficient to depolarize completely the presynaptic membrane [28]. Figure 2a shows that rising KCl up to 150 mM elicited an additional increase of fluorescence. Therefore, this concentration was used in our subsequent experiments.

Influence of glucose withdrawal on DiSC3(5) fluorescence. a Kinetics of DiSC3(5) fluorescence increase after KCl addition. KCl was added, when indicated. Curves represent eight independent experiments. b Influence of hypoglycemia on DiSC3(5) fluorescence. Con—control, GW—glucose withdrawal; synaptosomes were incubated 30 min at 37 °C in medium without glucose. R− synaptosomes were incubated 30 min at 37 °C in medium without glucose and in the presence of 10 µM rotenone. R + O synaptosomes were incubated 30 min at 37 °C in medium without glucose and in the presence of 10 µM rotenone + oligomycin (5 µg/mL). Bars represent DiSC3(5) fluorescence increase within 4 min after addition of 150 mM KCl. Data presented are mean values ± SEM of at least 6 experiments. 100 % level corresponds to fluorescence increase in glucose-containing control medium. *P ≤ 0.05 versus 100 %. **P ≤ 0.01 versus 100 %. Also **P ≤ 0.01 rotenone versus rotenone + oligomycin
Figure 2b shows that glucose deprivation decreased the dye uptake by 21 %.
This result shows that hypoglycemia reduces potassium-induced plasma membrane depolarization. It is suggested that glucose deprivation depolarizes plasma membrane in rat brain synaptosomes.
The addition of mitochondrial toxin rotenone alone or rotenone + oligomycin in glucose-free medium additionally lowered the dye content as much as 28 or 59 %, respectively.
There is statistically significant difference between membrane potentials of synaptosomes incubated in glucose-free medium in the presence rotenone and rotenone + oligomycin (P ≤ 0.01) (Fig. 2b). Rotenone alone was more effective (Fig. 2b).
Influence of Glucose Removal from Incubation Medium on Synaptic Vesicle Recycling
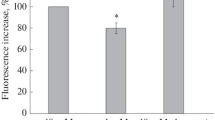
Figure 3a shows that 100 nM bafilomycin A1, an inhibitor V-ATPase, was able to increase the acridine orange fluorescence intensity. Glucose deprivation did not change the dye uptake by synaptic vesicles (Fig. 3b). This result shows that hypoglycemia does not change synaptic vesicle membrane depolarization induced by bafilomycin. It is suggested that glucose deprivation does not change pH gradient in synaptic vesicles.

Influence of glucose withdrawal on acridine orange fluorescence. a Kinetics of fluorescence increase after bafilomycin (100 nM) addition. Bafilomycin (b) was added, when indicated. Curve represents five independent experiments. b Influence of hypoglycemia on acridine orange fluorescence increase evoked by bafilomycin. Con—control, GW—glucose withdrawal; synaptosomes were incubated 30 min at 37 °C in medium without glucose. Bars represent acridine orange fluorescence increase within 5 min after addition of 100 nM of bafolomycin. Data presented are mean values ± SEM of 5 experiments. 100 % level corresponds to fluorescence increase in glucose-containing control medium. c Kinetics of fluorescence increase after KCl (60 mM) addition. KCl was added, when indicated. Curves represent nine independent experiments. Con—control, GW—glucose withdrawal; synaptosomes were incubated 30 min at 37 °C in medium without glucose. d Influence of hypoglycemia on acridine orange fluorescence increase evoked by KCl. Con—control, GW—glucose withdrawal; synaptosomes were incubated 30 min at 37 °C in medium without glucose. Bars represent maximal increase of acridine orange fluorescence after addition of 60 mM of KCl. Data presented are mean values ± SEM of 9 experiments. 100 % level corresponds to fluorescence increase in glucose-containing control medium. **P ≤ 0.01 versus 100 %
In contrast to membrane potential, synaptic vesicle recycling is very sensitive to changes in solution osmolarity. It is well established that hypertonic shrinking leads to calcium-independent exocytosis in neurons and synaptosomes [29, 30]. To avoid possible contribution of hypertonic-induced exocytosis we choose 60 mM KCl less affecting the solution osmolarity than 150 mM KCl for investigation of the synaptic vesicle cycle in our experiments. Figure 3c shows typical biphase response of acridine orange to potassium-induced plasma membrane depolarization. The initial increase reflects exocytosis, and the subsequent decrease is mediated by endocytosis [14, 19]. Glucose deprivation abolished exocytotic response to KCl by 49 % (Fig. 3c, d).
Influence of Glucose Withdrawal from Incubation Medium on Mitochondrial pH Gradient
Figures 3a and 4a, b show that mitochondrial toxins as well as bafilomycin A1, an inhibitor V-ATPase, were capable of magnifying acridine orange fluorescence intensity. The difference between the two columns on Fig. 4b indicates that about 86 % of the dye is concentrated in synaptic vesicles and the remaining 14 % is concentrated in mitochondria. Glucose deprivation leads to a dramatic increase in acridine orange pool which is sensitive to mitochondrial toxins. Figure 4a, c show the threefold rise of dye’s response. Similarly, the response to proton ionophore CCCP was strongly increased (Fig. 4d, e). These data suggest that glucose deprivation increases pH gradient in intrasynaptosomal mitochondria.

Investigation of mitochondrial pH gradient. a Kinetics of fluorescence increase after rotenone (10 µM) + oligomycin (5 µg/mL) addition. Oligomycin (O) and rotenone (R) were added, when indicated. Curves represent five independent experiments. Con—control, GW—glucose withdrawal; synaptosomes were incubated 30 min at 37 °C in medium without glucose. b Increase of acridine orange fluorescence after additions of bafilomycin (Baf) and rotenone + oligomycin (R + O). Bars represent acridine orange fluorescence increase within 5 min after addition of 100 nM of bafilomycin (100 nM) or 10 µM rotenone + 5 µg/mL oligomycin. Data presented are mean values ± SEM of at least 5 experiments. **P ≤ 0.01 versus zero. c Influence of glucose withdrawal on rotenone + oligomycin evoked acridine orange fluorescence increase. Con—control, GW—glucose withdrawal; synaptosomes were incubated 30 min at 37 °C in medium without glucose. Bars represent acridine orange fluorescence increase within 5 min after addition of 10 µM rotenone + 5 µg/mL oligomycin. Data presented are mean values ± SEM of 5 experiments. 100 % level corresponds to fluorescence increase in glucose-containing control medium. *P ≤ 0.05 versus 100 %. d Kinetics of fluorescence increase after CCCP (10 µM) addition. CCCP was added, when indicated. Curves represent six independent experiments. Con—control, GW—glucose withdrawal; synaptosomes were incubated 30 min at 37 °C in medium without glucose. e Influence of glucose withdrawal on CCCP evoked acridine orange fluorescence increase. Con—control, GW—glucose withdrawal; synaptosomes were incubated 30 min at 37 °C in medium without glucose. Bars represent acridine orange fluorescence increase within 5 min after addition of 10 µM CCCP. Data presented are mean values ± SEM of 6 experiments. 100 % level corresponds to fluorescence increase in glucose-containing control medium. **P ≤ 0.01 versus 100 %
Discussion
In the present paper, we investigated the influence of hypoglycemia on the plasma membrane potential, pH gradient of synaptic vesicles and membrane potential of intrasynaptosomal mitochondria. The major finding from our results is that presynaptic terminal functionality is preserved for 30 min without complete dissipation of ion gradients upon depriving energy substrates (Figs. 1, 2, 3). This observation is further supported by the results of Morgenthaler et al. [31], who reported that synapses of cortical neurons were functional for at least 10 min without glucose supplementation. Relatively minor depolarization of the plasma membrane and the mitochondrial membrane was also found under glucose deprivation in guinea pig synaptosomes [7]. The same study found reduced but not completely depleted ATP pool [7]. If synapses are able to survive for a certain time without being supplemented with external energy substrates, what do they consume to maintain ion transport? Our experiments using mitochondrial toxins provided clear evidences of the demand of presynaptic terminals for functional mitochondria in order to maintain the plasma membrane potential without glucose (Fig. 2b). Rotenone alone yielded more significant depolarization compared to rotenone + oligomycin (Fig. 2b). This finding is thought to be explained by reversion of mitochondrial ATPase and the beginning of cytosolic ATP hydrolysis to maintain the mitochondrial potential in case of electron-transport chain failure [11, 22]. Nonetheless, the small difference between these two conditions precludes the use of the synaptic cytosolic ATP pool as an energy depot (Fig. 2b). Concentrations of rotenone and oligomycin applied were sufficient to fully dissipate the intrasynaptosomal mitochondria potential [26]. It is established that synaptosomal cytosol contains fatty acid, organic acid and amino acids which can be used as nonglycolitic energy substrates to fuel mitochondria [32]. In our experiments, this pool is most likely consumed to hold the membrane potentials.
Next, we compared the effects of hypoglycemia on different potentials. The susceptibility of examined parameters to a hypogycemic state was as follows: mitochondrial potential > plasma membrane potential > pH gradient in synaptic vesicles (Figs. 1, 2, 3). Surprisingly, glucose deprivation had virtually no impact on the latter parameter (Fig. 3b). Dissipation of potential in synaptic vesicles was expected would be facilitated first of all due to higher surface/volume ratio. The maintenance of the pH gradient is important for preventing neurotransmitter leakage from synaptic vesicles followed by their release through reversion of plasma membrane transporters [16–18]. This implies that main biophysical characteristics of neuronal presynaptic endings do not promote the development of excitotoxicity under a hypoglycemic state.
Our results suggest that calcium-dependent exocytosis is the process mostly sensitive to energy deficit (Fig. 3c, d). Mechanistically, three underlying causes can be proposed. These are the decrease of pH gradients generated by V-ATPase, depletion of synaptic vesicles due to exocytosis provoked by plasma membrane depolarization (Fig. 2a), and impairment of ATP-dependent priming due to the energy deficit [33, 34]. Our results clearly rule out the first possibility (Fig. 3b). We have shown that glucose deprivation led to the decrease of DiSC3(5) uptake by 21 % (Fig. 2b) that could not be considered a strong depolarization. We have previously demonstrated the positive shift of synaptosomal plasma membrane potential having comparable amplitude, which has been induced by activation of presynaptic glycine receptors, was not sufficient to overcome the threshold for initiation of exocytosis [21]. Therefore, priming failure is the most probable mechanism underlying exocytosis inhibition.
Acridine orange are widely used to monitor the recycling and pH gradient of synaptic vesicles [14, 17, 19, 35]. This fluorescent dye is concentrated in acidic organelles wherein it is quenched by protonation. When a pH gradient is dissipated, e.g. during exocytosis, acridine orange is dequenched with a concomitant rise of fluorescence [14]. Inside synaptic vesicles, the pH value is about 5.5 [36]. Therefore, synaptic vesicles are supposed to be the primary, if not the only, compartment for localization of acridine orange in synaptosomes [14, 35]. However, in human ECV304 cells, the pH value was reported to be about 6.88 in the intermembrane mitochondrial space, about 7.59 in cytosol, and about 7.78 in mitochondrial matrix [36]. Acidification of the intermembrane space is mediated by the electron transport chain as rotenone + oligomycin were able to eliminate completely this effect [37]. Figures 3a, 4a, b showed that bafilomycin and rotenone + oligomycin accelerated the acridine orange fluorescence in synaptosomes, although mitochondrial response was significantly less pronounced. In our experimental settings, the dye reacted to mitochondrial toxins and has apparently been located in mitochondrial intermembrane spaces. This approach can be used for monitoring pH gradients in intrasynaptosomal mitochondria. Out of a total content, about 15 % of dye molecules are distributed in the mitochondria according to our estimation (Fig. 4b). However, glucose deprivation leads to more than a threefold increase of this pool indicating strong reinforcement of mitochondrial pH gradients (Fig. 4a, c). A proton ionophore was able to dissipate the pH gradient in synaptic vesicles [13, 38] and in mitochondria [40, 41]. Therefore, it is not surprising that proton ionophore CCCP induced an increase of acridine orange fluorescence in synaptosomes [18, 35]. We have shown that under a hypoglycemic state the CCCP-induced acridine orange response was also increased in as much as threefold (Fig. 4d, e). In view of the fact, that glucose deprivation had no impact on bafilomycin-induced dye response (Fig. 3b), this finding would support the idea of an increase of the mitochondrial pH gradient between matrix and intermembrane space.
Our results suggest that hypoglycemia leads to the increase of ΔpH in mitochondria (Fig. 4). The mechanism inducing this process is not clear. It is established that starvation causes mitochondrial membrane remodeling and resculpting of mitochondria morphology [42]. Probably, the increase of ΔpH might be a consequence of these changes. The increase of the pH gradient is thought to be a compensatory mechanism which enables maintaining the function of mitochondrial ATPase in unfavorable conditions.
Synaptic vesicles have membrane potential and pH gradient as do mitochondria [43]. It is well established that transport of different neurotransmitters can be regulated by changes in ΔΨ and ΔpH of synaptic vesicle membranes via different ways [13, 43, 44]. Therefore, changes in pH gradient may result in modification of ion-transporting capability of mitochondria, for instance calcium transport [12].
Summarizing, our results confirms that the main biophysical characteristics of neuronal presynaptic terminals do not promote the non-controlled release of neurotransmitters in the case of acute glucose lowering in blood. Such neuronal processes as inhibition of exocytosis, increase of the mitochondrial pH gradient and preservation of the vesicular pH gradient can serve as protective mechanisms.
References
Isaev NK, Stel’mashuk EV, Zorov DB (2007) Cellular mechanisms of brain hypoglycemia. Biochemistry (Moscow) 72:471–478
Suh SW, Hamby AM, Swanson RA (2007) Hypoglycemia, brain energetic, and hypoglycemic neuronal death. Glia 55:1280–1286
Languren G, Montiel T, Julio-Amilpas A, Massieu L (2013) Neuronal damage and cognitive impairment associated with hypoglycemia: an integrated view. Neurochem Int 63:331–343
Auer RN, Olsson Y, Siesjo BK (1984) Hypoglycemic brain injury in the rat. Correlation of density ob brain damage with the EEG isoelectric time: a quantitative study. Diabetes 33:1090–1098
Lau A, Tymianski M (2010) Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch 460:525–542
Wieloch T (1985) Hypoglycemia-induced neuronal damage prevented by an N-methyl-d-aspartate antagonists. Science 230:681–683
Kauppinen RA, Nicholls DG (1986) Synaptosomal bioenergetics. The role of glycolisis, pyruvate oxidation and responses to hypoglycaemia. Eur J Biochem 158:159–165
Silver IA, Deas J, Erecinska M (1997) Ion homeostasis in brain cells: differences in intracellular ion responses to energy limitation between cultured neurons and glial cells. Neuroscience 78:589–601
Suh SW, Aoyama K, Chen Y, Garnier P, Matsumori Y, Gum E, Liu J, Swanson RA (2003) Hypoglycemic neuronal death and cognitive impairment are prevented by poly(ADP-ribose) inhibitors administered after hypoglycemia. J Neurosci 23:10681–10690
Kann O, Kovacs R (2007) Mitochondria and neuronal activity. Am J Physiol Cell Physiol 292:C641–657
Chinopoulos C, Adam-Vizi V (2010) Mitochondria as ATP consumers in cellular pathology. Biochem Biophys Acta 1802:221–227
Rizzuto R, De Stefani D, Raffaello A, Mammucari C (2012) Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol 13:566–578
Maycox PR, Deckwerth T, Hell JW, Jahn R (1988) Glutamate uptake by brain synaptic vesicles. Energy dependence of transport and functional reconstitution in proteoliposomes. J Biol Chem 263:15423–15428
Zoccarato F, Cavallini L, Alexandre A (1999) The pH-sensitive dye acridine orange as a tool to monitor exocytosis/endocytosis in synaptosomes. J Neurochem 72:625–633
Wilhelm BG, Mandad S, Truckenbrodt S, Kröhnert K, Schäfer C, Rammner B, Koo SJ, Claβen GA, Krauss M, Haucke V, Urlaub H, Rizzoli SO (2014) Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science 344:1023–1028
Sulzer D, Chen TK, Lau YY, Kristensen H, Rayport S, Ewing A (1995) Amphetamine redistributes dopamine from synaptic vesicles to the cytosol and promotes reverse transport. J Neurosci 15:4102–4108
Tarasenko AS, Storchak LG, Himmelreich NH (2008) Alpha-latrotoxin affects mitochondrial potential and synaptic vesicles proton gradient of nerve terminals. Neurochem Int 52:392–400
Lemeshchenko VV, Pekun TG, Vasim TV, Fedorovich SV (2012) Y-27632 induces calcium-independent glutamate release in rat brain synaptosomes by the mechanism which is distinct from exocytosis. Biofizika 57:454–459 (in Russian)
Waseem TV, Rakovich AA, Lavrukevich TV, Konev SV, Fedorovich SV (2005) Calcium regulates the mode of exocytosis induced by hypotonic shock in isolated neuronal presynaptic endings. Neurochem Int 46:235–242
Waseem TV, Kolos VA, Lapatsina LP, Fedorovich SV (2007) Hypertonic shrinking but not hypotonic swelling increases sodium concentration in rat brain synaptosomes. Brain Res Bull 73:135–142
Waseem TV, Fedorovich SV (2010) Presynaptic glycine receptors influence plasma membrane potential and glutamate release. Neurochem Res 35:1188–1195
Chinopoulos C, Tretter L, Adam-Vizi V (1999) Depolarization of in situ mitochondria due to hydrogen peroxide-induced oxidative stress in nerve terminals: inhibition of α-ketoglutarate dehydrogenase. J Neurochem 73:220–228
Hajos F (1975) An improved method for the preparation of synaptosomal fractions in high purity. Brain Res 93:485–489
Pekun TG, Lemeshchenko VV, Lyskova TI, Waseem TV, Fedorovich SV (2013) Influence of intra- and extracellular acidification on free radical formation and mitochondria membrane potential in rat brain synaptosomes. J Mol Neurosci 49:211–222
Lowry O, Rosenbrough H, Farr H, Randall R (1951) Protein measurements with Folin reagent. J Biol Chem 193:265–279
Fedorovich SV (2013) Piracetam induces plasma membrane depolarization in rat brain synaptosomes. Neurosci Lett 553:206–210
Pekun TG, Waseem TV, Fedorovich SV (2014) Depolarization of plasma membrane of rat brain synaptosomes at extra- and intracellular acidification. Biophysics 59:77–80
Blaustein MP, Goldring MP (1975) Membrane potentials in pinched-off presynaptic nerve terminals monitored with a fluorescent probe: evidence that synaptosomes have potassium diffusion potentials. J Physiol 247:589–615
Rosenmund C, Stevens CF (1996) Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron 16:1197–1207
Waseem TV, Lapatsina LP, Fedorovich SV (2008) Influence of integrin-blocking peptide on gadolinium- and hypertonic shrinking-induced neurotransmitter release in rat brain synaptosomes. Neurochem Res 33:1316–1324
Morgenthaler FD, Kraftsik R, Catsikas S, Magistretti PJ, Chatton J-Y (2006) Glucose and lactate are equally effective in energizing activity-dependent synaptic vesicle turnover in purified cortical neurons. Neuroscience 141:157–165
Lee DY, Xun Z, Platt V, Budworth H, Canaria CA, McMurray CT (2013) Distinct pools of non-glycolytic substrates differentiate brain regions and prime region-specific responses of mitochondria. Plos One 8:e68831
Südhof TC (2013) Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 80:675–690
Imig C, Min SW, Krinner S, Arancillo M, Rosenmund C, Südhof TC, Rhee J, Brose N, Cooper BH (2014) The morphological and molecular nature of synaptic vesicle priming at presynaptic active zones. Neuron 84:416–431
Krisanova N, Sivko R, Kasatkina L, Borysov A, Borisova T (2014) Excitotoxic potential of exogenous ferritin and apoferritin: changes in ambient level of glutamate and synaptic vesicle acidification in brain nerve terminals. Mol Cell Neurosci 58:95–104
Van der Kloot W (2003) Loading and recycling of synaptic vesicles in the Torpedo electric organ and the vertebrate neuromuscular junction. Prog Neurobiol 71:269–303
Melnik VI, Bikbulatova LS, Gulyaeva NV, Bazyan AS (2001) Synaptic vesicle acidification and exocytosis studied with acridine orange fluorescence in rat brain synaptosomes. Neurochem Res 26:549–554
Porcelli AM, Ghelli A, Zanna C, Pinton P, Rizzuto R, Rugolo M (2005) pH difference across the outer mitochondrial membrane measured with a green fluorescent protein mutant. Biochem Biophys Res Commun 326:799–804
Wolosker H, de Souza DO, de Meis L (1996) Regulation of glutamate transport into synaptic vesicles by chloride and proton gradient. J Biol Chem 271:11726–11731
Tretter L, Chinopoulos C, Adam-Vizi V (1998) Plasma membrane depolarization and disturbed Na+ homeostasis induced by the protonophore carbonyl cyanide-p-trifluoromethoxyphenyl-hydrazon in isolated nerve terminals. Mol Pharmacol 53:734–741
Alekseenko AV, Lemeshchenko VV, Pekun TG, Waseem TV, Fedorovich SV (2012) Glutamate-induced free radical formation in rat brain synaptosomes is not dependent on intrasynaptosomal mitochondria membrane potential. Neurosci Lett 513:238–242
Patten DA, Wong J, Khacho M, Soubanner V, Mailloux RJ, Pilon-Larose K, MacLaurin JG, Park DS, McBride HM, Trinkle-Mulcahy L, Harper M-E, Germain M, Slack RS (2014) OPA1-dependent cristae modulation is essential for cellular adaptation to metabolic demand. EMBO J 33:2676–2691
Edwards RH (2007) The neurotransmitter cycle and quantal size. Neuron 55:835–858
Hell JW, Maycox PR, Jahn R (1990) Energy dependence and functional reconstitution of the γ-aminobutyric acid carrier from synaptic vesicles. J Biol Chem 265:2111–2117
Acknowledgments
This work was supported by Belorussian Republican Foundation of Basic Investigation (Grant B13-066). Foundation body had no involvement in study design, in the collection, analysis and interpretation of data, in the writing of the report, and in the decision to submit the article for publication.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Hrynevich, S.V., Pekun, T.G., Waseem, T.V. et al. Influence of Glucose Deprivation on Membrane Potentials of Plasma Membranes, Mitochondria and Synaptic Vesicles in Rat Brain Synaptosomes. Neurochem Res 40, 1188–1196 (2015). https://doi.org/10.1007/s11064-015-1579-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-015-1579-0