
Abstract
Mammalian AMP-activated protein kinase (AMPK) functions as a metabolic switch. It is composed of 3 different subunits and its activation depends on phosphorylation of a threonine residue (Thr172) in the α-subunit. This phosphorylation can be brought about by 5-aminoimidazole-4-carboxamide 1-β-d-ribofuranoside (AICAR) which in the cells is converted to a monophosphorylated nucleotide mimicking the effect of AMP. We show that the preparation of cultured astrocytes used for metabolic studies expresses AMPK, which could be phosphorylated by exposure of the cells to AICAR. The effect of AMPK activation on glutamate metabolism in astrocytes was studied using primary cultures of these cells from mouse cerebral cortex during incubation in media containing 2.5 mM glucose and 100 µM [U-13C]glutamate. The metabolism of glutamate including a detailed analysis of its metabolic pathways involving the tricarboxylic acid (TCA) cycle was studied using high-performance liquid chromatography analysis supplemented with gas chromatography–mass spectrometry technology. It was found that AMPK activation had profound effects on the pathways involved in glutamate metabolism since the entrance of the glutamate carbon skeleton into the TCA cycle was reduced. On the other hand, glutamate uptake into the astrocytes as well as its conversion to glutamine catalyzed by glutamine synthetase was not affected by AMPK activation. Interestingly, synthesis and release of citrate, which are hallmarks of astrocytic function, were affected by a reduction of the flux of glutamate derived carbon through the malic enzyme and pyruvate carboxylase catalyzed reactions. Finally, it was found that in the presence of glutamate as an additional substrate, glucose metabolism monitored by the use of tritiated deoxyglucose was unaffected by AMPK activation. Accordingly, the effects of AMPK activation appeared to be specific for certain key processes involved in glutamate metabolism.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Brain function is energetically demanding and hence, a continuous supply of energy is needed [1]. This may be achieved by regulatory mechanisms capable of maintaining an optimal energy supply at proper time and place. It is also important to control energy expenditure, especially during periods of limited energy supply and, therefore, regulatory pathways balancing anaplerotic and cataplerotic functions with regard to cellular energy status are vital. A strict control of the pathways involved in metabolism of the excitatory neurotransmitter glutamate is likewise essential for normal brain function. In addition to being the most abundant neurotransmitter in the brain glutamate is a key metabolite in the coupling of cellular energy- and amino acid metabolism. Glutamate is released from glutamatergic neurons and interacts with receptors at the post-synaptic terminal of the receiving neuron. Astrocytes surrounding the synapses are essential in order to terminate neurotransmission since the majority of glutamate is taken up and metabolized by astrocytes [2]. Glutamate is an important substrate to support energy metabolism in astrocytes either via complete oxidative degradation or by providing a net synthesis of tricarboxylic acid (TCA) cycle intermediates that increases the capacity for glucose oxidation [3, 4].
The energy sensor, mammalian AMP-activated protein kinase (AMPK) has been described as the master metabolic switch in various cell types [5]. AMPK is composed of three different subunits; a catalytic α-subunit and regulatory β- and γ-subunits, with each subunit existing in different isoforms [6–8]. The sensor can be activated by ADP as well as AMP, when either of these molecules binds to the γ-subunit of AMPK. This binding promotes phosphorylation by upstream kinases at a conserved threonine residue (Thr172) located in the α-subunit of AMPK. In the case of energy imbalances, e.g. due to the lack of energy substrates or hypoxia, peripheral cells compensate by initiating several mechanisms, including the phosphorylation, i.e. activation of AMPK [9, 10]. Activated AMPK down-regulates anabolic processes such as glycogen production, and fatty acid and cholesterol synthesis in order to reduce the ATP expenditure thereby preventing depletion of ATP. Furthermore, the activated sensor initiates a number of catabolic processes, such as glucose uptake, glycolysis, and fatty acid oxidation [8, 11, 12].
Astrocytes are important metabolic partners for neurons as they express the anaplerotic enzyme pyruvate carboxylase (PC), which is not present in the neurons [13]. This enzyme is quantitatively the most important anaplerotic enzyme in brain [14] and hence responsible for de novo synthesis of glutamate from glucose. The lack of PC in neurons necessitates metabolic interaction between astrocytes and neurons in order to maintain glutamatergic signaling since released glutamate is transported into astrocytes as stated above. Subsequent to uptake, glutamate can be amidated to glutamine catalyzed by the astrocyte specific enzyme glutamine synthetase [15]. Astrocytes release glutamine as precursor for synthesis of neurotransmitter glutamate in neurons as part of the glutamate-glutamine cycle [16]. As an alternative to conversion to glutamine, glutamate taken up by astrocytes may enter oxidative metabolism in the TCA cycle via α-ketoglutarate. α-Ketoglutarate can be formed from glutamate by transamination catalyzed primarily by aspartate aminotransferase (AAT), or by oxidative deamination via the action of glutamate dehydrogenase (GDH) [2, 3]. The oxidative deamination leads to an elevation of the total amount of TCA cycle intermediates, which catalytically facilitates glucose oxidation. A complete oxidative degradation of glutamate requires pyruvate recycling, which is initiated by the action of malic enzyme (ME) catalyzing the oxidative decarboxylation of malate to pyruvate. An alternative route includes the concerted action of phosphoenolpyruvate carboxykinase and pyruvate kinase. However, the activity of ME has been shown to be much higher than that of phosphoenolpyruvate carboxykinase in cultured astrocytes [17], and therefore only pathways involving ME will be considered.
Metabolism of glucose and glutamate in astrocytes is highly interconnected. However, the regulation of these metabolic pathways is poorly understood, particularly the role of AMPK signaling. In order to investigate this, cultured neocortical astrocytes were exposed to the AMP-analogue 5-aminoimidazole-4-carboxamide 1-β-d-ribofuranoside (AICAR) in the presence of [U-13C]glutamate. AICAR is intracellularly converted to the monophosphorylated nucleotide ZMP and mimicks the effects of AMP without changing the ATP/AMP or ATP/ADP ratio in the cell. AICAR is capable of activating AMPK both allosterically and by facilitating phosphorylation [5, 18, 19]. Effects of AMPK activation on the metabolic fate of the glutamate carbon skeleton were studied in detail using 13C-labeled glutamate and gas chromatography coupled to mass spectrometry (GC–MS). The effect of AMPK activation on glucose uptake and phosphorylation in the presence of glutamate as additional substrate was determined using tracer amounts of the radioactively labeled glucose analogue, 2-deoxyglucose, a procedure developed by Sokoloff et al. [20].
Using this analytical approach we found that activation of AMPK had profound effects on metabolic handling of glutamate in astrocytes. The entrance of glutamate into the TCA cycle was reduced but neither its cellular uptake nor the conversion to glutamine was affected. The flux of glutamate derived carbon through the reactions catalyzed by ME and PC was reduced which affected synthesis of releasable citrate, a process which is normally prominent in astrocytes [21, 22].
Materials and Methods
Materials
NMRI mice (7-day-old) were obtained from Harlan (The Netherlands) and housed in the animal facility at Department of Drug Design and Pharmacology, University of Copenhagen (Copenhagen, Denmark) prior to isolation of brain tissue. Plasticware for cell culturing was acquired from NUNC A/S (Roskilde, Denmark). Dulbecco’s modified Eagle’s Medium (DMEM) powder, N-methyl-N-(tert-butyldimethylsilyl)trifluoroacetamide (MTBSTFA) and N,N-dimethylformamide (DMF) were purchased from Sigma-Aldrich (St. Louis, MO, USA) while 5-aminoimidazole-4-carboxamide 1-β-d-ribofuranoside (AICAR) was bought from Tocris Bioscience (Bristol, UK). Foetal calf serum (FCS) was manufactured by Gibco, Life Technologies (Carlsbad, CA, USA). 2-[1,2-3H]-Deoxy-d-glucose was obtained from PerkinElmer (Boston, MA, USA) and L-[U-13C]glutamic acid (97–99 % enriched) was manufactured by Cambridge Isotope Laboratories (Andover, MA, USA). HPLC and GC–MS chemicals and columns were purchased from Agilent Technologies (Santa Clara, CA, USA) and Phenomenex (Torrance, CA, USA), respectively. Pierce BCA protein assay-kit was produced by Thermo Fischer Scientific (Rockford, IL, USA). xCell Surelock NuPAGE system, buffers and chemicals for the western blots were purchased from Life Technologies, while a complete protease inhibitor cocktail was purchased from Roche Applied Science (Mannheim, Germany), Immobilon-P polyvinylidene fluoride (PVDF) membrane of 0.45 µm pore size was purchased from Millipore (Billerica, MA, USA). Enhanced chemiluminescence (ECL) chemicals as well as rabbit anti-β-actin antibody (1:5000) were manufactured by Abcam (Cambridge, UK). Rabbit anti- phospho-AMPKα (Thr172) antibody (#2531) and rabbit anti-AMPKα antibody (total AMPKα; #2532) were bought from Cell Signalling (Leiden, The Netherlands) and goat-anti-rabbit horseradish peroxidase (HRP)-conjugated secondary antibody from Dako (Glostrup, Denmark). All other chemicals were obtained in purest grade from regular commercial suppliers.
Primary Neocortical Mouse Astrocyte Cultures
Culture medium: Reconstituted DMEM powder was supplemented with 26.2 mM NaHCO3, 2.5 mM glutamine, 6 mM glucose, 100 i.u/mL penicillin, 0.04 mM phenol red and FCS in suitable concentration and sterile filtered. Primary cultures of cortical astrocytes were prepared from mouse brains as described by Hertz et al. [23] and Walls et al. [24]. In brief, the neocortices were dissected from 7-day-old mice. The brain tissue was squeezed through a nylon filter (pore size of 80 μM) into a DMEM-solution containing 20 % FCS. To dissociate the tissue, the homogenate was processed 3 times with a 20 mL syringe mounted with a steel cannula (13 G × 5″). Subsequently, the cell suspension was seeded in 25 cm2 tissue culture flasks at the density of approximately 0.2 mouse brain cortex/flask or as indicated below and kept at 37 °C in a humidified CO2-incubator (95 % atmospheric air/5 % CO2). The cell culture medium was changed twice a week with a gradual reduction of FCS (1st week: 20 %, 2nd week: 15 %, 3rd week: 10 %). In the 3rd week dibuturyl-cAMP was added to the culture medium in order to differentiate the cells. After 3 weeks of culturing the cells were used for experiments. The cell culture medium was changed to a corresponding fresh medium 1 day prior to the experiments. On the day of the experiments the cultures were examined microscopically to evaluate the confluency and the morphological appearance of the cells.
Westen Blot
The cells were grown in 6-well-plates at the density of 1.8 cortices/plate according to the culturing protocol provided above. One day prior to the experiment the media were exchanged in order to replenish nutrients. On the day of the experiment the media were removed and the cells incubated for 60 min at 37 °C in basic DMEM supplemented with 2.5 mM glucose in the presence or absence of 0.5 mM AICAR. At the end of the incubation period the media were removed and the cells washed with ice-cold phosphate buffered saline (PBS, 137 mM NaCl, 2.7 mM KCl, 7.3 mM Na2HPO4, 1.5 mM, KH2PO4, pH 7.4). The cells were then harvested in 2 % SDS (sodium dodecyl sulphate) prepared in PBS and supplemented with a protease inhibitor cocktail. Protein content in the prepared lysates was measured using the Pierce BCA protein assay according to manufacturer’s instructions.
Proteins were denatured by boiling the samples at 95 °C for 5 min in the NuPAGE sample buffer supplemented with the NuPAGE reducing agent. The gels were loaded with 18 μg protein per lane and the proteins were resolved on NuPAGE Bis–Tris 4–12 % pre-cast gels (200 V, 1 h). Proteins were transferred onto a PVDF membrane for 1 h at 30 V in the NuPAGE transfer buffer supplemented with 20 % methanol. The membranes were then blocked for 2 h in 5 % BSA solution in TBS-T (50 mM Tris, 100 mM NaCl, pH 7.2, 0.05 % Tween-20) at room temperature. The membranes were incubated overnight with either anti-AMPKα total or anti-phospho-AMPKα (Thr172) antibody (4 °C; 1:1000 dilution in 5 % BSA in TBS-T), followed by 1.5 h incubation with anti-rabbit IgG conjugated with horse radish peroxidase (room temperature; 1:2000 dilution in 5 % BSA in TBS-T). The immuno-reactive bands were visualized using the ECL development method. In order to avoid stripping of the antibodies, two identically loaded gels were always resolved and processed simultaneously. One of them was used for AMPKα total and the other for phospho-AMPKα (Thr172) staining. Both stainings were corrected for β-actin content present in the same sample lane. ImageJ [25] was used for semiquantification of the ECL signal. In short, intensity of each band was expressed as the percent of the total intensity of the particular staining on a gel. Thus, determined intensity of either AMPKα total or phospho-AMPKα (Thr172) was then divided by the percent intensity of the corresponding β-actin stain for the sample lane in question. Subsequently, the corrected AMPKα-phospho (Thr172) was divided by the corrected AMPKα-total signal. These values were used for subsequent statistical analysis.
Incubation with [U-13C]glutamate
The astrocytes were grown in 25 cm2 flask following the dissection and isolation protocol provided above. On the day of the experiment DMEM-medium (without glutamine) containing 100 μM L-[U-13C]glutamate and 2.5 mM glucose ± 0.5 mM AICAR was CO2-equilibrated (95 % atmospheric air/5 % CO2) immediately before the experiment was performed. AICAR was also added to the cell cultures 30 min prior to the initiation of the incubation. The media in the cultures were discarded, the cells rinsed twice with PBS containing 0.9 mM CaCl2 and 0.5 mM MgCl2, and 3.5 mL of incubation medium was added. Subsequently, the cultures were incubated for 2 h at 37 °C in a CO2-incubator (95 % atmospheric air/5 % CO2). After 1 h the cultures were spiked with 100 μM L-[U-13C]glutamate to ensure a concentration of minimum 100 μM L-glutamate. After 2 h the medium was collected and the cells were extracted with 70 % ethanol. Afterwards the tubes were centrifuged (20 min, 20,000×g, 4 °C) in order to separate the protein (pellet) from the extract (supernatant). The extracts and media were lyophilized, reconstituted in 250 and 400 μL double-distilled water, respectively, and used for subsequent mass spectrometric- and HPLC-analyses.
Quantitative Determination of Amino Acids
The amounts of relevant amino acids in the extracts (aspartate, glutamate, serine, glutamine, alanine, valine, leucine and isoleucine) and media (glutamine and alanine) were determined using reverse phased HPLC with LC-10ADVP liquid chromatograph coupled to an RF-10AXL fluorescent detector (Shimadzu). Pre-column derivatization with o-phthaldialdehyde (OPA) and fluorescent measurement (excitation λ = 350 nm, emission λ = 450 nm) were performed. For separating the amino acids an Agilent Eclipse AAA column (4.6 mm × 150 mm, pore size 5 μm) with a mobile phase gradient based on mobile phase A and B was employed. Composition of mobile phase A: 0.02 M citrate, 0.06 M phosphate, pH 5.9 with 4.8 % acetonitrile. Composition of mobile phase B: 90 % acetonitrile. Mobile phase based gradient: from 4.5 to 16.5 min mobile phase B increased from 0 to 7 %, from 16.5 to 35 min from 7 to 50 % and from 36 to 38 min mobile phase B was reduced to 0 %. All of the amino acids of interest eluted within 32 min. The content of amino acids in extract and medium was calculated as nmol per culture, since the protein content of the cultures was not determined. However, we have no reason to believe that the exposure to AICAR and activation of AMPK for 2.5 h induces detectable changes in protein content.
Metabolic Mapping of Metabolites of Interest Using Gas Chromatography Coupled to Mass Spectrometry (GC–MS)
Extract- and media samples were centrifuged (20 min, 20,000×g, 4 °C), adjusted to pH 1–2 with HCl and evaporated to complete dryness under nitrogen flow. Organic extraction was performed with 96 % ethanol/benzene followed by derivatization with 14 % DMF/86 % MTBSTFA, a procedure modified after Mawhinney et al. [26]. Standards containing unlabeled metabolites of interest, cell extracts and media were analyzed using a gas chromatograph (Agilent Technologies 7820A chromatograph, J&W GC column HP-5MS, parts no. 19091S-433) coupled to a mass spectrometer (Agilent Technologies 5977E). The isotopic enrichment of the metabolites of interest were corrected for natural abundance of 13C using the unlabeled standards and calculated according to Bieman [27]. The data are presented as % labeling of M + X, where M is the mass of the unlabeled molecule and X is the number of labeled C-atoms in the metabolite.
2-Deoxyglucose Uptake
The cells were grown in 24-well-plates at the density of 1.0 cortices/plate according to the culturing protocol provided above. Incubation medium containing 2.5 mM glucose and 100 μM glutamate was CO2-equilibrated with 95 % atmospheric air/5 % CO2 just before the experiment. AICAR (0.5 mM) was added 30 min prior to the initiation of incubation. The media were discarded and the astrocytes were rinsed twice with pre-warmed PBS containing 0.9 mM CaCl2 and 0.5 mM MgCl2 followed by the addition of 300 μL incubation medium. The cultures were incubated for 15 min in a 37 °C CO2-incubator (95 % atmospheric air/5 % CO2) and 2-[1,2-3H]deoxyglucose was added to a final radioactivity of 3.2 µCi/mL. The cultures were incubated for further 15 min in a 37 °C CO2-incubator (95 % atmospheric air/5 % CO2) and the incubations were terminated by collecting the medium and washing the cells twice with ice-cold PBS. The cells were harvested and dissolved in 0.1 M KOH overnight (4 °C). The cell suspensions were neutralized with 10 % (v/v) formic acid, mixed with Ecoscint A and the radioactivity determined using a liquid scintillation counter (Tri-Carb, 2900TR Perkin Elmer, Waltham, MA, USA).
Statistics
In testing differences between the control and the AICAR-treated astrocytes, unpaired Student’s t-test was conducted using GraphPad Prism 6.0 software. The data are presented as averages ± standard error of the mean. Statistically significant differences were detected with p values < 0.05.
Results
AMPK α-Subunit Expression
In order to determine the degree of AMPK expression and activation in the murine primary astrocyte cultures, western blot analysis was performed (Fig. 1a, b). The determination of the total amount of AMPK α-subunit shows that the astrocytes express considerable total amounts of AMPKα. The low background phosphorylation of Thr172 in the AMPKα subunit was increased when the cells were incubated with 0.5 mM AICAR for 60 min. This analysis clearly shows that astrocytic AMPKα is capable of responding to the pharmacological AMPK activator, AICAR, which was found to increase AMPK phosphorylation approximately 3-fold. The use of 2.5 mM AICAR had no additional effect on the extent of phosphorylation of AMPK (results not shown).

Representative immunoblots (a) of astrocytes incubated for 60 min in DMEM supplemented with 2.5 mM glucose in the absence (−) or presence (+) of the AMPK activator—AICAR (0.5 mM). The upper panel shows the blot probed for phospho-AMPKα (Thr172), while the lower panel represents the immunoblot probed for the total AMPKα subunit. All lanes were loaded with 18 µg protein. Results of the semi-quantitative analysis (b) of the induction of AMPKα phosphorylation. The immunoblots were densitometrically analysed in ImageJ. The relative signal intensity of AMPKα total and phospho-AMPKα (Thr172) was corrected each for their corresponding β-actin signal. Finally, the corrected phospho-AMPKα (Thr172) value was divided by the corrected AMPKα total intensity for each experimental sample (n = 3). Values are shown ± SEM; *p < 0.05 (unpaired Student’s t-test)
Glutamate Metabolism
Astrocyte cultures were incubated in medium containing [U-13C]glutamate (100 μM) and glucose (2.5 mM), with or without 0.5 mM AICAR activating AMPK. Subsequent to the incubation, the extracts and media were analyzed using HPLC and GC–MS in order to measure quantitative amounts of metabolites and determine 13C-labeling, respectively. The labeling pattern provides information about the effects of AMPK activation on the particular metabolic pathways involved in metabolism of glutamate. Quantification of total amounts of selected amino acids was performed in cell extracts as well as in media using HPLC. The presence of AICAR during the incubation of the cultured astrocytes in a medium containing glutamate and glucose had no effect on the intra- or extracellular levels of the selected amino acids (Table 1).
Incubations performed in medium containing [U-13C]glutamate gave rise to a high labeling (%) in glutamate (M + 5) (Fig. 2a). No significant differences in intracellular M + 5 isotopomers of glutamate and glutamine between control and AICAR-treated astrocytes were detected (Fig. 2a, b). This indicates that activating AMPK does not affect the two essential components of the glutamate-glutamine cycle, namely glutamate uptake and glutamine synthesis catalyzed by glutamine synthetase.

Control (black columns) and AICAR treated (grey columns) astrocytes were incubated for 2 h in basic DMEM supplemented with [U-13C]glutamate (100 µM) and glucose (2.5 µM)as detailed in “Materials and Methods”. Cell extracts (a) and media (c) were analyzed using GC–MS for determination of the percentage distribution of 13C-labeled metabolites that arise from direct metabolism of [U-13C]glutamate in the TCA cycle (b). Effects of AICAR treatment were tested using unpaired Student’s t-test and an asterisk indicates a statistically significant difference (*p < 0.05). Values are averages ± SEM, (n = 5–12). Each experimental condition consists of 3–5 samples derived from 3 individual cell culture batches. [U-13C]Glutamate (M + 5 Glu) is via direct metabolism converted to [U-13C]glutamine (M + 5 Gln) or it is entering the TCA cycle for conversion into TCA cycle intermediates such as α-[U-13C]ketoglutarate (M + 5 α-KG), [U-13C]succinate (M + 4 Suc), [U-13C]fumarate (M + 4 Fum) and [U-13C]malate (M + 4 Mal). [U-13C]Aspartate (M + 4 Asp) is formed due to the equilibrium with [U-13C]oxaloacetate catalyzed by aspartate aminotransferase (a, b). [U-13C]Oxaloacetate condenses with unlabeled acetyl-CoA and [3,4,5,6-13C]citrate (M + 4 Cit) is formed. Glutamine (M + 5) and citrate (M + 4) are released and detected in the medium (b, c)
[U-13C]Glutamate is taken up into the mitochondria via either one of the glutamate carriers (GC1 & GC2) or aralar, and 13C label can enter the TCA cycle via conversion of glutamate to α-ketoglutarate (M + 5). α-Ketoglutarate (M + 5) is decarboxylated to succinyl-CoA and further metabolized to succinate (M + 4), M + 4 fumarate, malate and oxaloacetate (Fig. 2b). Aspartate (M + 4) is in equilibrium with M + 4 oxaloacetate due to the high activity of AAT. A significant reduction in the labeling (%) of α-ketoglutarate (M + 5), succinate (M + 4) and aspartate (M + 4) of 30, 51 and 11 %, respectively, was observed in AICAR-treated astrocytes compared to control cells (Fig. 2a). Since the amount and the labeling of intracellular [U-13C]glutamate was not affected by activation of AMPK, these reductions of labeling in TCA cycle intermediates indicate that either the transport of glutamate into the mitochondria or the entry of glutamate via α-ketoglutarate into the TCA cycle is down regulated by the activation of AMPK signaling. The reduction in labeling (%) of fumarate (M + 4), malate (M + 4) and citrate (M + 4) was not statistically significant, but tended to be reduced (p values: 0.09, 0.11, 0.18, respectively) (Fig. 2a).
Citrate (M + 4) is formed from condensation of M + 4 oxaloacetate and unlabeled acetyl-CoA (Fig. 2b). Citrate as well as glutamine are released by the astrocytes to the medium (Fig. 2b). A 25 % reduced labeling (%) of citrate (M + 4) in the medium was seen in AICAR-treated astrocytes compared to control cells whereas that of glutamine (M + 5) was unaffected (Fig. 2c). As observed intracellularly, the labeling (%) of released glutamine (M + 5) was not affected by the presence of AICAR (Fig. 2c).
Metabolism Via Malic Enzyme, Pyruvate Carboxylase, Lactate- and Pyruvate-Dehydrogenase
Glutamate (M + 5) may, when entering the TCA cycle, be converted to malate (M + 4) which, in turn, can be decarboxylated to pyruvate (M + 3) via the action of ME (Fig. 3b). Pyruvate (M + 3) may be reduced to lactate (M + 3) catalyzed by lactate dehydrogenase (LDH) or transaminated into alanine (M + 3) catalyzed by alanine aminotransferase (ALAT). As can be seen in Fig. 3b, astrocytes release lactate (M + 3) and alanine (M + 3) to the media. A statistically significant decrease (29 %) in the labeling of lactate (M + 3) released to the media was observed when comparing AICAR-treated astrocytes with control cells (Fig. 3a). In contrast, the labeling of extracellular alanine was unaffected (Fig. 3a). The amount of lactate in the medium primarily produced from unlabeled glucose was estimated from the GC–MS measurements and the amount of lactate released was not affected by AMPK activation (results not shown).

Control (black columns) and AICAR treated (grey columns) astrocytes were incubated for 2 h in basic DMEM supplemented with [U-13C]glutamate (100 µM) and glucose (2.5 µM) as detailed in “Materials and Methods”. Media (a, d) and cell extracts (c) were analyzed using GC–MS for determination of the percentage distribution of 13C labeling in intracellular metabolites that arise from metabolism of [U-13C]glutamate via malic enzyme (ME) combined with pyruvate carboxylase (PC) and/or pyruvate dehydrogenase activity, i.e. pyruvate recycling (b). Effects of AICAR treatment were tested using unpaired Student’s t test and an asterisk indicates a statistically significant difference (*p < 0.05). Values are averages ± SEM, (n = 5–12). Each experimental condition consists of 3-5 samples derived from three individual cell culture batches. [U-13C]Malate (M + 4 Mal) formed from direct metabolism of [U-13C]glutamate (M + 5 Glu) may be decarboxylated to [U-13C]pyruvate (M + 3 Pyr) catalyzed by ME (b). M + 3 Pyr may be reduced to [U-13C]lactate (M + 3 Lac) or transaminated by alanine aminotransferase into [U-13C]alanine (M + 3 Ala) that is subsequently released to the medium (a, b). M + 3 Pyr may be carboxylated by the action of PC forming triple labeled oxaloacetate (M + 3) being in equilibrium with triple labeled aspartate (M + 3 Asp). Oxaloacetate (M + 3) is in equilibrium with Mal, Fum and Suc, all being M + 3. M + 3 Oxaloacetate may condense with either unlabeled acetyl-CoA forming triple labeled citrate (M + 3 Cit) or double labeled acetyl-CoA (b) forming quintuple labeled citrate (M + 5 Cit). Alternatively, uniformly labeled citrate (M + 6 Cit) may be formed from condensation of double labeled acetyl-CoA and [U-13C]oxaloacetate (M + 4) formed from direct metabolism of M + 5 Glu (b)
Instead of being reduced to lactate or transaminated to alanine, pyruvate may be carboxylated to oxaloacetate by the anaplerotic enzyme PC, located in the mitochondria. Pyruvate can furthermore be oxidatively decarboxylated to acetyl-CoA by pyruvate dehydrogenase (PDH) (Fig. 3b). Pyruvate (M + 3) decarboxylated by PDH or carboxylated by PC gives rise to either acetyl-CoA (M + 2) or oxaloacetate (M + 3), respectively. Oxaloacetate (M + 3) may via back flux in the TCA cycle lead to M + 3 labeling in malate, fumarate and succinate (Fig. 3b). If oxaloacetate (M + 3) condenses with M + 2 or unlabeled acetyl-CoA, citrate (M + 5) or (M + 3) is formed. Alternatively, if oxaloacetate (M + 4) (direct synthesis) condenses with acetyl-CoA (M + 2), citrate (M + 6) is formed. Furthermore, oxaloacetate (M + 3) can be transaminated in the AAT catalyzed reaction giving rise to aspartate (M + 3) (Fig. 3b). We observed statistically significant reductions with regard to M + 3 labeling in succinate, fumarate, malate and aspartate (43, 27, 31 and 31 %, respectively) in AICAR-treated astrocytes compared to control cells (Fig. 3c). This observation points to a reduction of flux of glutamate derived carbon via ME and/or PC when AMPK is active. The labeling of intracellular citrate (M + 3, M + 5 and M + 6) was not significantly affected (p-values: 0.079, 0.064, 0.97, respectively) but that of the corresponding extracellular pool of citrate (M + 3, M + 5) was reduced (Fig. 3d). Glutamate (M + 4) is a result of pyruvate recycling, and it includes condensation of [1,2-13C]acetyl-CoA and oxaloacetate (M + 3) formed by the concerted action of ME and PC. Glutamate (M + 4) labeling was 9 % reduced in AICAR-treated astrocytes compared to control cells (Fig. 3c), which likely reflects the reduction of oxaloacetate (M + 3) labeling formed by pyruvate carboxylation. The reductions in extracellular citrate (M + 5) labeling also likely originate from reduction in oxaloacetate (M + 3) labeling. This is supported by the fact that the relative reduction observed in aspartate (M + 3) labeling is higher than that observed in citrate (M + 5) and glutamate (M + 4) (Fig. 3). Interestingly, labeling of citrate (M + 5) in the media was higher than that observed in the extracts (p = 0.0043) (Fig. 3c, d). From this observation it may be inferred that citrate, the synthesis of which is coupled to the PC reaction is preferentially exported to the media. In addition to that, AMPK affects a compartment with extensive PC activity and in which citrate is synthesized from glutamate. No statistically significant changes in citrate (M + 6) were detected in the media.
Uptake and Phosphorylation of 2-Deoxyglucose
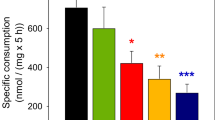
2-Deoxyglucose is a glucose analogue, which is taken up and phosphorylated by hexokinase but cannot undergo further metabolism, causing it to be trapped intracellularly as 2-deoxyglucose-6-phosphate. 2-Deoxyglucose mimics glucose with regard to the transmembrane transport and phosphorylation and by using a tracer amount of 2-[3H]deoxyglucose it is possible to monitor glucose uptake and phosphorylation [20]. According to Sokoloff et al. [20] the accumulated radioactivity in the cells reflects total uptake and metabolism of glucose regardless of the end product of glucose metabolism. We observed no statistically significant difference in glucose uptake and phosphorylation in the presence of glutamate when comparing astrocytes incubated with AICAR to control cells (Fig. 4).

Uptake and phosphorylation of 2-[3H]deoxyglucose in cultured astrocytes in the absence (black column) or presence (grey column) of AICAR. The experimental condition is described in “Materials and Methods”. Values representing counts per culture are averages ± SEM (n = 9) derived from 3 individual cell culture batches. Effects of AICAR treatment was tested using unpaired Student’s t-test showing no statistically significant difference
Discussion
AMPK is described as the major metabolic switch in peripheral cells, which regulates several mechanisms involved in energy homeostasis [5, 8]. As reviewed in detail by Ronnett et al., the presence of AMPK in hypothalamic energy-sensing neurons supports the function as a master regulator of energy homeostasis, affecting food intake, energy expenditure and body weight [28]. In neurons and skeletal muscle cells, translocation of the glucose transporters, GLUT3 and GLUT4 to the plasma membrane is increased by AMPK activation [29, 30]. In astrocytes, glucose is the major energy substrate and building block for de novo synthesis of glutamate and glutamine [1, 31] and AMPK activation has been shown to induce synthesis of the major activator of glycolysis, fructose-2,6-bisphosphate, thereby promoting astrocytic survival when the nitric oxide (NO) level is elevated [28, 32]. NO also induced inhibition of mitochondrial respiration, but that effect was not shown to be linked to the activation of AMPK [32]. In the brain, astrocytes surround synapses and take up and metabolize glutamate as energy substrate, in addition to glucose [2]. In the presence of glutamate, activation of AMPK had no significant effect on glucose uptake in astrocytes. The main glucose transporter in astrocytes is GLUT1 [33, 34]. GLUT1 has been shown to translocate to the plasma membrane in response to AMPK activation in cardiac myocytes [35]. The presence of glutamate as an additional and efficient energy substrate may be the reason why AMPK activation does not induce an increased glucose uptake in the cultured astrocytes.
Glutamate-glutamine Cycle Versus TCA Cycle Metabolism of Glutamate
In astrocytes, glutamate is either converted into glutamine for release or it enters the mitochondria to support metabolism in the TCA cycle. Activation of AMPK did not lead to changes in intracellular glutamate and intra- and extracellular glutamine (M + 5) labeling and amounts. This suggests that the essential parts of the glutamate–glutamine cycle, i.e. glutamate uptake, glutamine synthesis and release are not affected by AMPK activation, although glutamate transport and synthesis of glutamine are energy requiring processes (see Schousboe et al. [2] ). In contrast, the entrance of glutamate into the TCA cycle was decreased as observed by a reduced α-ketoglutarate (M + 5) labeling. It may be speculated that the processes located in astrocytes, which are needed to maintain neuronal signaling, are prioritized compared to energy production. It should be noted that in the present study, AMPK is activated in the presence of glucose as energy substrate, which means that the astrocytes are unlikely to have an impaired ATP production. The reduced labeling of α-ketoglutarate, which is also reflected in subsequent TCA cycle intermediates, may originate from lower uptake of glutamate into the mitochondria or a lower conversion of glutamate to α-ketoglutarate, a prerequisite for TCA cycle metabolism of the carbon skeleton of glutamate. This entry of glutamate into the TCA cycle requires either GDH or transaminase activity. However, GDH is thought to be the main enzyme responsible for conversion of glutamate into α-ketoglutarate in astrocytes [36–38]. Interestingly, one study in hypoxia tolerant crayfish, has shown a reduction in allosteric activation of GDH by ADP when AMPK was activated [39].
Malic Enzyme Coupled to Pyruvate Carboxylase, Pyruvate Dehydrogenase or Lactate Dehydrogenase
As reviewed by Sonnewald [40] anaplerosis via PC in astrocytes must be coupled to cataplerosis (i.e., exit of carbon atoms from the TCA cycle) at a matching magnitude because the TCA cycle cannot function as a carbon-sink [40]. Cataplerosis of TCA cycle intermediates requires the concerted action of ME and PDH, i.e. pyruvate recycling. We found no statistically significant reduction in the labeling of malate (M + 4) which is formed via direct metabolism of [U-13C]glutamate but that of lactate (M + 3) was reduced suggesting that AMPK activation may lower the flux of glutamate derived carbon through the ME and LDH catalyzed reactions. However, since the labeling of alanine (M + 3) was unchanged, the effect is likely restricted to the LDH reaction. It should be kept in mind that the LDH as well as ME catalyzed reactions are sensitive to the redox state of the cell, which may be related to the activation of AMPK. Distinct labeling of the lactate and alanine pool may point to intracellular compartmentation of the pyruvate pool, a phenomenon that has been observed repeatedly [21, 41].
Lactate formation from glutamate is a partial oxidation and degradation of the carbon skeleton and since lactate is released it is an eventual loss of metabolite, thus a catabolic pathway that only partially supports energy production. A reduction in the flux to lactate from glutamate may support an anaplerotic function of glutamate that increases the capacity for glucose oxidation during AMPK activation. Pyruvate may be oxidatively decarboxylated to acetyl-CoA via PDH, i.e. pyruvate recycling, instead of being converted to lactate. AMPK activation does not seem to affect the flux through this pathway since the labeling patterns primarily reflect the alterations of flux in the reaction catalyzed by PC.
The decarboxylation of malate to pyruvate catalyzed by ME followed by carboxylation of pyruvate to oxaloacetate seems redundant. Nevertheless, the labeling of the TCA cycle intermediates and aspartate clearly shows that the glutamate derived carbon is metabolized through this pathway. The effect of AMPK activation on this pathway was even more prominent than that observed on direct metabolism of glutamate in the TCA cycle. Particularly, the flux of glutamate derived pyruvate via the anabolic process pyruvate carboxylation was down regulated in astrocytes exposed to AMPK activation.
Citrate Synthesis and Release
Citrate is extensively synthesized and released from cultured astrocytes and is found in a high concentration in cerebrospinal fluid [22, 42]. Citrate is not efficiently taken up by neurons and is therefore not a candidate to support neither neuronal glutamate synthesis nor energy metabolism [42]. However, it has been suggested that extracellular citrate regulates the concentration of Zn2+-ions by chelation thereby modulating the excitability of neurons [43]. This marked release of citrate from astrocytes is known to be coupled to an extensive pyruvate carboxylation [21]. Addition of K+ and bicarbonate stimulates pyruvate carboxylation as well as citrate release in astrocytes [42, 44]. In the present study the activation of AMPK led to reduced pyruvate carboxylation of glutamate derived pyruvate and this was clearly reflected in the labeling of extracellular citrate. This confirms that releasable citrate is synthesized from oxaloacetate in a cellular compartment exhibiting extensive PC activity [21]. AMPK activation is known to down regulate anabolic processes [45] and these results suggest that glutamate derived carbon is important for the anabolic synthesis of releasable citrate and that this pathway is regulated by AMPK. The higher labeling of citrate, extracellularly compared to intracellularly supports that glutamate is preferentially supporting synthesis of the releasable pool of citrate. The two intracellular pools of citrate, i.e. the one representing the main intracellular pool and the one from which citrate is released are compartmentalized as previously shown [21]. It may be postulated that the reduced entrance of glutamate into the TCA cycle observed during AMPK activation is due to the fact that glutamate serves an anabolic role for the TCA cycle, i.e. being precursor for citrate synthesis.
In conclusion, the effects of AMPK activation appeared to be specific for certain processes involved in glutamate metabolism, i.e. entrance of glutamate into the TCA cycle and synthesis of releasable citrate via pyruvate carboxylation. Since AMPK activation induces catabolic processes these results may suggest an anabolic function of glutamate in the TCA cycle. In contrast, processes of the glutamate-glutamine cycle, i.e. glutamate uptake, glutamine synthesis and release were not affected by AMPK activation.
Abbreviations
- AAT:
-
Aspartate aminotransferase
- AICAR:
-
5-Aminoimidazole-4-carboxamide 1-β-d-ribofuranoside
- ALAT:
-
Alanine aminotransferase
- AMPK:
-
AMP activated protein kinase
- BCA:
-
Bicinchoninic acid
- BSA:
-
Bovine serum albumin
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- FCS:
-
Foetal calf serum
- GDH:
-
Glutamate dehydrogenase
- GC–MS:
-
Gas chromatography–mass spectrometry
- HPLC:
-
High-performance liquid chromatography
- LDH:
-
Lactate dehydrogenase
- ME:
-
Malic enzyme
- OPA:
-
o-Phthaldialdehyde
- PBS:
-
Phosphate buffered saline
- PC:
-
Pyruvate carboxylase
- PDH:
-
Pyruvate dehydrogenase
- TCA:
-
Tricarboxylic acid
References
Hertz L, Dienel GA (2002) Energy metabolism in the brain. Int Rev Neurobiol 51:1–102
Schousboe A, Scafidi S, Bak LK, Waagepetersen HS, McKenna MC (2014) Glutamate metabolism in the brain focusing on astrocytes. Adv Neurobiol 11:13–30. doi:10.1007/978-3-319-08894-5_2
McKenna MC (2013) Glutamate pays its own way in astrocytes. Front Endocrinol 4:191. doi:10.3389/fendo.2013.00191
Dienel GA (2013) Astrocytic energetics during excitatory neurotransmission: what are contributions of glutamate oxidation and glycolysis? Neurochem Int 63(4):244–258. doi:10.1016/j.neuint.2013.06.015
Winder WW, Hardie DG (1999) AMP-activated protein kinase, a metabolic master switch: possible roles in type 2 diabetes. Am J Physiol 277(1 Pt 1):E1–10
Mitchelhill KI, Stapleton D, Gao G, House C, Michell B, Katsis F, Witters LA, Kemp BE (1994) Mammalian AMP-activated protein kinase shares structural and functional homology with the catalytic domain of yeast Snf1 protein kinase. J Biol Chem 269(4):2361–2364
Stapleton D, Gao G, Michell BJ, Widmer J, Mitchelhill K, Teh T, House CM, Witters LA, Kemp BE (1994) Mammalian 5’-AMP-activated protein kinase non-catalytic subunits are homologs of proteins that interact with yeast Snf1 protein kinase. J Biol Chem 269(47):29343–29346
Hardie DG, Ross FA, Hawley SA (2012) AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13(4):251–262. doi:10.1038/nrm3311
Salt IP, Johnson G, Ashcroft SJ, Hardie DG (1998) AMP-activated protein kinase is activated by low glucose in cell lines derived from pancreatic beta cells, and may regulate insulin release. Biochem J 335(Pt 3):533–539
Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, Van den Berghe G, Carling D, Hue L (2000) Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol 10(20):1247–1255
Kemp BE, Mitchelhill KI, Stapleton D, Michell BJ, Chen ZP, Witters LA (1999) Dealing with energy demand: the AMP-activated protein kinase. Trends Biochem Sci 24(1):22–25
Moore F, Weekes J, Hardie DG (1991) Evidence that AMP triggers phosphorylation as well as direct allosteric activation of rat liver AMP-activated protein kinase. A sensitive mechanism to protect the cell against ATP depletion. Eur J Biochem/FEBS 199(3):691–697
Yu AC, Drejer J, Hertz L, Schousboe A (1983) Pyruvate carboxylase activity in primary cultures of astrocytes and neurons. J Neurochem 41(5):1484–1487
Patel MS (1974) The relative significance of CO2-fixing enzymes in the metabolism of rat brain. J Neurochem 22(5):717–724
Norenberg MD, Martinez-Hernandez A (1979) Fine structural localization of glutamine synthetase in astrocytes of rat brain. Brain Res 161(2):303–310
Bak LK, Schousboe A, Sonnewald U, Waagepetersen HS (2006) Glucose is necessary to maintain neurotransmitter homeostasis during synaptic activity in cultured glutamatergic neurons. J Cereb Blood Flow Metab 26(10):1285–1297. doi:10.1038/sj.jcbfm.9600281
Cruz F, Scott SR, Barroso I, Santisteban P, Cerdan S (1998) Ontogeny and cellular localization of the pyruvate recycling system in rat brain. J Neurochem 70(6):2613–2619
Corton JM, Gillespie JG, Hawley SA, Hardie DG (1995) 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem 229(2):558–565
Zhang L, Frederich M, He H, Balschi JA (2006) Relationship between 5-aminoimidazole-4-carboxamide-ribotide and AMP-activated protein kinase activity in the perfused mouse heart. Am J Physiol Heart Circ Physiol 290(3):H1235–H1243. doi:10.1152/ajpheart.00906.2005
Sokoloff L, Reivich M, Kennedy C, Des Rosiers MH, Patlak CS, Pettigrew KD, Sakurada O, Shinohara M (1977) The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J Neurochem 28(5):897–916
Waagepetersen HS, Sonnewald U, Larsson OM, Schousboe A (2001) Multiple compartments with different metabolic characteristics are involved in biosynthesis of intracellular and released glutamine and citrate in astrocytes. Glia 35(3):246–252
Sonnewald U, Westergaard N, Krane J, Unsgard G, Petersen SB, Schousboe A (1991) First direct demonstration of preferential release of citrate from astrocytes using [13C]NMR spectroscopy of cultured neurons and astrocytes. Neurosci Lett 128(2):235–239
Hertz L, Juurlink B, Hertz E, Fosmark H (1989) Preparation of primary cultures of mouse (rat) astrocytes. In: Shahar A, De Vellis J, Haber B (eds) A dissection and tissue culture manual of the nervous system. Liss Inc, New York, pp 105–108
Walls A, Bak L, Sonnewald U, Schousboe A, Waagepetersen H (2014) Metabolic mapping of astrocytes and neurons in culture using stable isotopes and gas chromatography–mass spectrometry (GC-MS). In: Hirrlinger J, Waagepetersen HS (eds) Brain energy metabolism, vol 90. Neuromethods. Springer New York, pp 73–105. doi:10.1007/978-1-4939-1059-5_4
Schneider CA, Rasband WS, Eliceiri KW (2012) NIH image to imagej: 25 years of image analysis. Nat Methods 9(7):671–675
Mawhinney TP, Robinett RS, Atalay A, Madson MA (1986) Analysis of amino acids as their tert.-butyldimethylsilyl derivatives by gas-liquid chromatography and mass spectrometry. J Chromatogr 358(1):231–242
Biemann K (1962) Organic chemistry applications. Mass spectrometry. McGraw, New York, pp 223–227
Ronnett GV, Ramamurthy S, Kleman AM, Landree LE, Aja S. (2009) AMPK in the brain: its roles in energy balance and neuroprotection. J Neurochem 109 (Suppl 1):17–23.
Kurth-Kraczek EJ, Hirshman MF, Goodyear LJ, Winder WW (1999) 5′ AMP-activated protein kinase activation causes GLUT4 translocation in skeletal muscle. Diabetes 48(8):1667–1671
Weisova P, Concannon CG, Devocelle M, Prehn JH, Ward MW (2009) Regulation of glucose transporter 3 surface expression by the AMP-activated protein kinase mediates tolerance to glutamate excitation in neurons. J Neurosci 29(9):2997–3008. doi:10.1523/JNEUROSCI.0354-09.2009
Hertz L, Peng L, Dienel GA (2007) Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J Cereb Blood Flow Metab 27(2):219–249. doi:10.1038/sj.jcbfm.9600343
Almeida A, Moncada S, Bolanos JP (2004) Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat Cell Biol 6(1):45–51. doi:10.1038/ncb1080
Barros LF, Bittner CX, Loaiza A, Ruminot I, Larenas V, Moldenhauer H, Oyarzun C, Alvarez M (2009) Kinetic validation of 6-NBDG as a probe for the glucose transporter GLUT1 in astrocytes. J Neurochem 109(Suppl 1):94–100. doi:10.1111/j.1471-4159.2009.05885.x
Simpson IA, Carruthers A, Vannucci SJ (2007) Supply and demand in cerebral energy metabolism: the role of nutrient transporters. J Cereb Blood Flow Metab 27(11):1766–1791. doi:10.1038/sj.jcbfm.9600521
Lee CT, Ussher JR, Mohammad A, Lam A, Lopaschuk GD (2014) 5′-AMP-activated protein kinase increases glucose uptake independent of GLUT4 translocation in cardiac myocytes. Can J Physiol Pharmacol 92(4):307–314. doi:10.1139/cjpp-2013-0107
Schousboe A, Westergaard N, Sonnewald U, Petersen SB, Huang R, Peng L, Hertz L (1993) Glutamate and glutamine metabolism and compartmentation in astrocytes. Dev Neurosci 15(3–5):359–366
Westergaard N, Drejer J, Schousboe A, Sonnewald U (1996) Evaluation of the importance of transamination versus deamination in astrocytic metabolism of [U-13C]glutamate. Glia 17(2):160–168. doi:10.1002/(SICI)1098-1136(199606)17:2<160:AID-GLIA7>3.0.CO;2-6
Yu AC, Schousboe A, Hertz L (1982) Metabolic fate of 14C-labeled glutamate in astrocytes in primary cultures. J Neurochem 39(4):954–960
Dawson NJ, Storey KB (2012) An enzymatic bridge between carbohydrate and amino acid metabolism: regulation of glutamate dehydrogenase by reversible phosphorylation in a severe hypoxia-tolerant crayfish. J Comp Physiol [B] 182(3):331–340. doi:10.1007/s00360-011-0629-4
Sonnewald U (2014) Glutamate synthesis has to be matched by its degradation: where do all the carbons go? J Neurochem 131(4):399–406. doi:10.1111/jnc.12812
Zwingmann C, Richter-Landsberg C, Leibfritz D (2001) 13C isotopomer analysis of glucose and alanine metabolism reveals cytosolic pyruvate compartmentation as part of energy metabolism in astrocytes. Glia 34(3):200–212
Westergaard N, Sonnewald U, Unsgard G, Peng L, Hertz L, Schousboe A (1994) Uptake, release, and metabolism of citrate in neurons and astrocytes in primary cultures. J Neurochem 62(5):1727–1733
Westergaard N, Banke T, Wahl P, Sonnewald U, Schousboe A (1995) Citrate modulates the regulation by Zn2+ of N-methyl-d-aspartate receptor-mediated channel current and neurotransmitter release. Proc Natl Acad Sci USA 92(8):3367–3370
Kaufman EE, Driscoll BF (1992) Carbon dioxide fixation in neuronal and astroglial cells in culture. J Neurochem 58(1):258–262
Hardie DG (2011) AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev 25(18):1895–1908. doi:10.1101/gad.17420111
Acknowledgments
This work was supported by the Novo Nordisk Foundation (2293). Heidi Nielsen and Anna Hansen are acknowledged for their excellent technical assistance. Stud. pharm. Omran Abdel Rahman is acknowledged for contributing to GC–MS analyses.
Author information
Authors and Affiliations
Corresponding author
Additional information
Special Issue: In honor of Dr. Gerald Dienel.
Rights and permissions
About this article

Cite this article
Voss, C.M., Pajęcka, K., Stridh, M.H. et al. AMPK Activation Affects Glutamate Metabolism in Astrocytes. Neurochem Res 40, 2431–2442 (2015). https://doi.org/10.1007/s11064-015-1558-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-015-1558-5