
Abstract
Gastrodin (GAS), an active constituent of the Chinese herbal medicine Tianma, has anti-oxidant and anti-inflammation activities but its protective effect to the prevention of neurotoxicity induced by ischemic stroke is unclear. In the present study, middle cerebral artery occlusion (MCAO) was used to establish a mice ischemic stroke model. Infarct volume ratio and neurobehavioral score were evaluated, Nissl staining was performed and the expression of cleaved Caspase 3, Bax and B cell lymphoma 2 (Bcl-2) were assessed at 24 h or 7 days after reperfusion. In addition, the total superoxide dismutase (SOD) activity and malondialdehyde (MDA) content, as well as the expression of Nuclear factor erythroid 2-related factor 2 (Nrf2), heme oxygenase-1 (HO-1), SOD1, phospho-Akt and total Akt and TNF-α and IL-1β in the ischemic hemispheres were also observed at 6 h after reperfusion to assess oxidative stress and inflammatory changes after GAS treatment. It was found that GAS, especially at high dose (100 mg/kg) reduced tested neuronal injury and neurobehavioral deficient in MCAO mice. Enhanced expression of cleaved Caspase 3 and Bax and decreased expression of Bcl-2 by MCAO were also reversed by GAS. Moreover, GAS treatment decreased the MDA content and the expression of TNF-α and IL-1β, and increased amount of SOD activity and the expression of HO-1 and SOD1 in GAS-treated ischemic brain. Furthermore, GAS significantly increased Akt phosphorylation and Nrf2 expression. These results support the neuroprotective effects of GAS, and the activation of Akt/Nrf2 pathway may play a critical role in the pharmacological action of GAS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Stroke is one of the primary causes of severe acquired disability worldwide [1]. The sudden blockage of blood flow from a thrombus or embolism causes about 80 % of all stroke victims experienced an ischemic event [2, 3]. Administration of the thrombolytic agent tissue plasminogen activator (tPA) is an efficient treatment and it is still the only FDA approved therapy for acute ischemic stroke [4, 5]. However, tPA has many shortcomings including the potential risk of hemorrhagic transformation, limited therapeutic window and efficacy [6, 7]. Thus, cerebral ischemia is still a major medical problem and novel and effective therapeutic approaches are still urgently required.
The exact mechanisms responsible for the brain damage suffering from ischemic insult are still not fully understood; there are increasing evidence suggesting that post-ischemia oxidative stress and inflammation are significant contributing factors to the pathogenic process [8–11]. Reactive oxygen species (ROS) are found greatly elevated which could exceed the antioxidant capacity of the ischemic brain, thus activate diverse signaling pathways and result in oxidative stress [12, 13], and ROS could initiate the expression of inflammatory cytokines [14], including tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β) and interleukin-6 (IL-6). Meanwhile, studies reported that systemic administration of antioxidants or anti-inflammatory agents could improve neurological deficit, reduce brain edema and infarct sizes and regulate cytokines expression in the cortex [15–17]. It suggests that agents with anti-oxidative or anti-inflammatory effects might be benefit for the treatment of cerebral ischemia.
Gastrodia elata Blume, known as tianma in Chinese, is a plant of the orchidaceae family, has been used for thousands of years in China. Pharmacological studies indicate that tianma has analgesic, nootropic, and anti-inflammatory effects, and improves microcirculation and general circulatory functions [18, 19]. The phenolic glucoside gastrodin (GAS) is a main phenolic compound derived from Gastrodia elata Blume root [20]. Studies indicate that GAS has a neuroprotective action against hypoxia in cultured cortical neuron [21], and GAS could inhibit the expression of inducible NO synthase (iNOS), cyclooxygenase-2 and pro-inflammatory cytokines in cultured LPS-stimulated microglia [22]. It may also improve learning ability and facilitate memory consolidation and retrieval [23]. Our previous study also showed that GAS could reduce the levels of IL-6 and IL-1β, down-regulate the expression of iNOS and inhibite p38 MAPK phosphorylation in the hippocampus of posttraumatic stress disorder model [24], and GAS could also reverse the expression of phospho-inhibitor of kappa B (p-iκB), nuclear factor-kappa B (NF-κB), and IL-1β of hippocampus in chronic unpredictable stress rats and protect hippocampal derived neural stem cell from IL-1β-induced damage [25], suggesting that GAS may suppress specific signaling pathways associated with the inflammatory response. Moreover, recent studies showed that the beneficial effects of gastrodin might result from its antioxidant properties [26]. GAS could protect primary cultured rat hippocampal neurons against amyloid-beta peptide-induced neurotoxicity and attenuate the reduction of catalase (CAT) and superoxide dismutase (SOD) expression via the activation of ERK1/2-Nrf2 pathway [27]. These observations lead to the hypothesis that GAS may provide neuroprotection against ischemic brain injury. To test this possibility, we investigated the potential neuroprotective efficacy of GAS in a mouse middle cerebral artery occlusion (MCAO) model. We also explored its anti-oxidative effects by assessing oxidative-related factors against stroke damage induced by MCAO in mice.
Materials and Methods
Experimental Animals and Drug Injection
The C57BL/6 J mice in this study were obtained from the Experimental Animal Center at the Fourth Military Medical University, Xi’an, China. The animals were housed at 21 ± 2 °C, with 60–70 % humidity for at least 1 weeks before surgery. They were also under a fixed 12-h light/dark cycle and had free access to food and water. Procedures for animals were reviewed and approved by the Ethics Committee for Animal Experimentation of the Fourth Military Medical University.
GAS (purity >99.2 %) was supplied by Kunming Pharmaceutical Corporation (Kunming, China). In the present study, GAS was dissolved in saline and administered intraperitoneally to all GAS groups at the same time. The doses were chosen according to previous pharmacological studies of GAS in rats [28, 29] and adjusted into dose for mice according to our preliminary tests. After the adaptive phase, mice were randomly divided into five groups: sham (only received saline), MCAO + vehicle (saline), MCAO + GAS (L) (low-dose, L = 10 mg/kg), MCAO + GAS (M) (medium dose, M = 50 mg/kg), MCAO + GAS (H) (high-dose, H = 100 mg/kg). Saline or GAS were immediately injected intraperitoneally at the onset of cerebral reperfusion.
Induction of Transient Focal Cerebral Ischemia
The focal cerebral ischemia was induced by MCAO with an intraluminal filament as described previously [30, 31]. Briefly, mice were anesthetized with 2 % isoflurane carried by 2 L/min oxygen through a face mask. Six-0 monofilament nylon suture with a rounded tip (Beijing, shadong) was inserted through a small incision on the right common carotid artery and forwarded into the internal carotid artery until a small resistance was felt. And the filament was fixed with silk suture knot on the common carotid artery. After 1 h, the filament was slowly withdrawn to allow reperfusion. Regional cerebral blood (rCBF) flow was monitored using laser Doppler flowmetry (Perimed AB, PeriFlux System 5000, Stockholm, Sweden) in the ipsilateral cortex. MCAO was considered successful if rCBF sharply decreased below 30 % of baseline; successful reperfusion was considered if rCBF was recovered up to 80 % of baseline. Otherwise, animals were excluded from analysis and sacrificed by anesthesia death. The mice in sham group were not experienced middle cerebral artery occlusion; they just anesthetized with 2 % isoflurane and exposed their carotid artery. The insufficient ischemia rate is 15 %, and the premature death is 10 %.
Neurological Deficit Evaluation and Infarct Assessment
Neurological deficits were monitored 24 h or 7 days after vascular occlusion using the 6-point scoring system [32] by a blinded observer. Mice were scored as follows: zero, no apparent deficits; one, failure to extend left forepaw fully if pulled by the tail; two: left contralateral circling if pulled by the tail; three: circling or walking to the left; four: walking only if stimulated; five: unresponsiveness to stimulation and with depressed level of consciousness. Immediately after neurological deficit evaluation, mice were decapitated and brains were dissected into six equal thickness coronal slices (1 mm per slice) and immediately stained with 2 % 2,3,5-triphenyltetrazolium chloride (TTC) (Sigma-Aldrich, St Louis, MO, USA, T8877) at 37 °C for 10 min. The infarct volumes were evaluated with the following formula: (total contralateral hemispheric volume − total ipsilateral hemispheric stained volume)/total contralateral hemispheric volume × 100.
Nissl Staining
At 24 h or 7 days after reperfusion, mice were subjected to deep anesthesia with 300 mg/kg i.p. chloral hydrate and perfused with saline followed by ice cold 4 % paraformaldehyde phosphate buffer through transcardiac perfusion. Brain tissues were then extracted and post-fixed in 4 % paraformaldehyde for 2 h at 4 °C. After successively infiltrated with 0.1 M PBS containing 20 and 25 % sucrose overnight, 10 μm coronal sections were cut with a cryostat (Leica, CM 1950) and stored at −20 °C until use. Nissl staining kit (Beyotime Institute of Biotechnology, China) was used to detect the numbers of morphologically normal neurons and neurons showing the features of ischemic cell change (shrunken cell bodies, triangulated, pyknotic nuclei, and eosinophilic cytoplasm) by two blinded investigators using ImagePro Plus 5.1 software (Media Cybernetics, Inc., Bethesda, MD). Neurons were counted in 10 different fields in each region in the cortex (primary somatosensory cortex, S1) and CA1 of hippocampus. The percentage of ischemic neurons was calculated through dividing the sum of ischemic neuron number by the total number of neurons in the 10 defined areas [33].
Measurement of Cerebral MDA and SOD Levels
Cerebral content of MDA, the product of lipid peroxidation, was measured according to the method by a detection kit purchased from Beyotime Institution of China. The absorbance of the supernatant was measured by spectrophotometry at 532 nm. Total cerebral SOD activity was determined using detection kits from Jiancheng Bio Institution, Nanjing, China, and the absorbance of the supernatant were measured by spectrophotometry at 450 nm.
Western Blotting
The ischemic hemispheres were lysed with SDS-PAGE sample buffer composed of 62.5 mM Tris–HCI, 2 % w/v SDS, 10 % glycerol, 50 mM DTT, and 0.1 % w/v bromphenol blue. After homogenization, extracts were clarified via centrifugation at 12,000g for 10 min. The supernatant was collected as the total cellular protein extract. Nuclear extracts were prepared using a NEPER Nuclear Extraction Kit (Pierce) according to manufacturer instructions. The protein concentrations were determined using a bicinchoninic acid protein assay kit (Pierce). Aliquots of the lysates with loading buffer (Beyotime Institution of China) were boiled for 10 min. and then cooled to room temperature. For western blot analysis, equal amounts of protein (20–40 mg) were separated with SDS polyacrylamide gel electrophoresis and blotted onto PVDF membranes (Millipore). Membrane was blocked with 5 % skim milk in Tris buffered saline (TBS) and then incubated overnight at 4 °C with the primary antibodies as follow: rabbit anti-Nrf2 antibody (1:200, Santa Cruz Biotechnology), rabbit anti-HO-1 antibody (1:500, Enzo Life Sciences), mouse anti-phospho-Akt antibody (1:1,000, Cell Signaling), rabbit anti-total Akt antibody (1:1,000, Cell Signaling), rabbit anti-cleaved caspase-3 antibody (1:1,000; Cell Signaling), rabbit anti-Bcl-2 antibody (1:2,000, Abcam) or rabbit anti-Bax antibody (1:2,500, Abcam), rabbit anti-IL-1β antibody (1:400, Abcam), goat anti-TNF-α antibody (Santa Cruz biotechnology, 1:500) and mouse anti-β-actin antibody. After washing, membranes were incubated with a horseradish peroxidase (HRP)-conjugated IgG secondary antibody in accordance with the origin of the primary antibody for 1 h at room temperature. Then the antibody-reactive bands were visualized on X-ray film using super ECL plus detection reagent (Thermo, USA, 34077). The protein quantifications were adjusted for the corresponding to β-actin level, which was not consistently changed by the different treatment conditions.
Statistical Analysis
The software SPSS 16.0 for Windows (SPSS Inc., Chicago, IL, USA) was used to conduct statistical analyses. All values, except for neurological scores, are presented as mean ± SEM and analyzed by one-way analysis of variance, and between-group differences were detected with the post hoc Student–Newman–Keuls test. The neurological deficit scores were expressed as median (range) and were analyzed with the Kruskal–Wallis test followed by the Mann–Whitney U test with the Bonferroni correction. Differences were considered significant when P < 0.05.
Results
GAS Reduced the Infarct Volume and Improved the Neurobehavioral Score Induced by Cerebral Ischemia
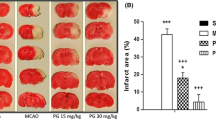
To detect the neuroprotective effect of GAS, mice were subjected to MCAO followed by GAS or vehicle treatment. As shown in Fig. 1, at 24 h after reperfusion, significant differences were found among groups in infarct volume ratio and neurobehavioral score (P < 0.01). Multiple comparisons further showed that MCAO induced definite infarct volume ratio and neurologic deficiency compared to sham group. These changes were alleviated by GAS at medium or high dose (P < 0.05 compared to MCAO + Vehicle). Additionally, high dose of GAS was more effective in alleviating ischemic brain injury for that compared to MCAO + GAS (L) group.

Gastrodin improved neurological scores and reduced infarct volumes 24 h after reperfusion injury. a A diagram of the experimental design for drug treatment and assessment of the ischemic injury. b Representative TTC-stained thick brain sections and quantitative evaluation of the infarction volume c and neurobehavioral score d for each group (n = 8/group). GAS significantly reduced infarct volumes (F 3, 24 = 8.847, P < 0.01) and improved neurological scores at 24 h after reperfusion. *P < 0.05, **P < 0.01; # P < 0.05 vs. MCAO + GAS (H) group, & P < 0.05vs.. MCAO + vehicle group
To detect if the neuroprotective effect of GAS was long lasting but not transient, mice were subjected to MCAO and then received GAS or vehicle once daily for 7 consecutive days. Infarct volume ratio and neurobehavioral score was detected at 7 days after reperfusion. As shown in Fig. 2, at 7 days after reperfusion, significant differences among groups were found for both infarct volume and neurobehavioral score (P < 0.01). Multiple comparisons showed that GAS at medium dose or high-dose alleviated the neuronal damage induced by MCAO, high dose of GAS was more potent in reducing cerebral damage induced by ischemia [P < 0.05 vs. MCAO + GAS (L)]. These results indicated that GAS dose-dependently reduced cerebral ischemic injury in mice.

GAS improved neurological scores and reduced infarct volumes 7 days after reperfusion injury. a The experimental design, b Representative TTC-stained thick brain sections, c quantitative evaluation of the infarction volume and d neurobehavioral score for each group (n = 8/group). GAS significantly reduced infarct volumes (F 3, 23 = 5.629, P < 0.01) and improved neurological scores 7 days after reperfusion. *P < 0.05, **P < 0.01; # P < 0.05 vs. MCAO + GAS (H) group, & P < 0.05 vs. MCAO + vehicle group
GAS Improved Morphology Damage and Inhibited Apoptosis Pathway in the Ischemic Hemispheres
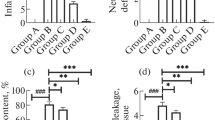
As shown in Fig. 3, at 24 h after reperfusion, there were significant differences between each group in the percentage of neuronal damage in the cortex and CA1 of hippocampus, as well as the protein expression of cleaved Caspase 3, Bax and Bcl-2. Post hoc comparisons further showed that MCAO induced significant neuron damage both in the cortex and CA1 hippocampus, increased the protein expression of cleaved Caspase 3 and Bax, but decreased the expression of Bcl-2 (P < 0.05). These changes were alleviated by GAS at medium dose or high-dose (P < 0.05). High dose of GAS further reduced the increase of Bax expression and inhibited the reduction of Bcl-2 expression caused by MCAO compared to MCAO + GAS (L) group.

GAS improved morphology damage and inhibited apoptosis pathway 24 h after reperfusion injury. Representative microphotographs a and quantitative evaluation showing Nissl staining to determine the cell damage by ischemia/reperfusion (I/R) in ischemic CA1 of hippocampus b and primary somatosensory cortex c of brain at 24 h after reperfusion. Scale bars = 100 μm. d Representative bands for each group. e, f, g Densitometric analysis for cleaved Caspase3, Bax and B-cell lymphoma 2 (Bcl-2), respectively. GAS relieved MCAO induced neuron damage both in the primary somatosensory cortex (F 4,35 = 10.330, P < 0.01) and CA1 of hippocampus (F 4,35 = 14.836, P < 0.01), and inhibited the increased expression of cleaved Caspase 3 (F 4,20 = 6.796, P < 0.01), Bax (F 4,20 = 16.676, P < 0.01) and Bcl-2 (F 4,20 = 7.681, P < 0.01) 24 h after reperfusion. *P < 0.05, **P < 0.01; # P < 0.05 vs. MCAO + GAS h group, & P < 0.05 vs. MCAO + vehicle group
As shown in Fig. 4, at 7 days after reperfusion, there were significant differences among groups for the percentage of injured neurons in the cortex and CA1 of hippocampus, as well as the protein expression of cleaved Caspase 3, Bax and Bcl-2. In accordance with the tendency of 24 h, Post hoc comparisons showed that medium dose or high-dose of GAS alleviated the neuronal damage induced by MCAO. Moreover, GAS at medium dose or high-dose inhibited the expression of cleaved Caspase 3 and Bax and elevated the expression of Bcl-2. Compared to MCAO + GAS (L) group, protein expression changes were more obvious in MCAO + GAS (H) group (P < 0.05), indicating that a larger dose of GAS was more effective in inhibiting pro-apoptotic protein expression and preserving anti-apoptotic protein expression.

GAS improved morphology damage and inhibited apoptosis pathway 7 days after reperfusion injury. Representative microphotographs a and quantitative evaluation showing Nissl staining to determine the cell damage by ischemia/reperfusion (I/R) in ischemic CA1 of hippocampus b and primary somatosensory cortex c of brain 7 days after reperfusion. Scale bars = 100 μm. d Representative bands for each group. e, f, g Densitometric analysis for cleaved Caspase3, Bax and Bcl-2, respectively. GAS relieved MCAO induced neuron damage both in the cortex (F 4,35 = 15.508, P < 0.01) and CA1 of hippocampus (F 4,35 = 21.126, P < 0.01), and inhibited the increased expression of cleaved Caspase 3 (F 4,20 = 14.405, P < 0.01), Bax (F 4,20 = 24.153, P < 0.01) and Bcl-2 (F 4,20 = 16.623, P < 0.01) after reperfusion for 7 days. *P < 0.05, **P < 0.01; # P < 0.05 vs. MCAO + GAS(H) group, & P < 0.05 vs. MCAO + vehicle group
GAS Reversed MCAO-Induced Oxidative Damage in the Ischemic Hemispheres
To evaluate the protective effect of GAS on the oxidative damage in mice brain induced by MCAO, we determined the protein expression of nuclear factor erythroid 2-related factor 2 (Nrf2), Heme oxygenase-1 (HO-1), SOD1, phospho-Akt, total Akt as well as the content of MDA and total cerebral SOD activity in the ischemic hemisphere at 6 h after reperfusion. As shown in Fig. 5, there were significant differences among groups in the expression of Nrf2, HO-1, SOD1 and phosphorylation level of Akt. The expression of SOD1 and phosphorylation level of Akt were significantly decreased after MCAO, which was dose dependently reversed by the administration of GAS. In addition, the expression of Nrf2 and HO-1 were significantly also dose dependently increased by the administration of GAS.

Modulation of Akt phosphorylation, Nrf2, HO-1 and SOD1 expression and MDA concentration and activity of SOD by GAS at 6 h after reperfusion injury. a A diagram of the experimental design. b Representative western blot bands and quantitative evaluation of Akt phosphorylation c, Nrf2 d, HO-1 e and SOD1 f. Histograms showed the concentration of MDA g and the activity of SOD1 h. GAS dependently reversed the increased expression of Nrf2 (F 4,20 = 9.974, P < 0.01), HO-1 (F 4,20 = 10.903, P < 0.01), SOD1 (F 4,20 = 13.479, P < 0.01) and phosphorylation level of Akt (F 4,20 = 10.672, P < 0.01), as well as the concentrations of malondialdehyde (MDA) (F 4,15 = 22.166, P < 0.01) and activity of SOD (F 4,15 = 9.636, P < 0.01) at 6 h after reperfusion. ## P < 0.05 vs. MCAO + GAS (H) and MCAO + GAS (M) group, # P < 0.05 vs. MCAO + GAS h group, *P < 0.05 vs. MCAO + vehicle group
There were also significant differences between each group in the concentrations of malondialdehyde (MDA) and activity of SOD. MCAO significantly increased the concentrations of MDA (Fig. 5g), which was product of lipid peroxidation, and the concentrations of MDA were dose-dependently reduced by the administration of GAS. In contrast, MCAO markedly decreased the activity of SOD compared with that of sham group. Different doses of GAS showed certain effect of increasing the activity of SOD (Fig. 5h).
GAS Inhibited the Expression of Inflammatory Cytokines in the Ischemic Hemispheres
To evaluate the anti-inflammatory effect of GAS in mice brain after MCAO, we determined the protein expression of TNF-α and IL-1β in the ischemic hemispheres at 6 h after reperfusion. As shown in Fig. 6, there were significant differences among groups in the expression of TNF-α and IL-1β. The expression of TNF-α and IL-1β were significantly increased in MCAO + Vehicle group, but this increase was inhibited by medium and/or high dose of GAS treatment.

Representative western blot bands and quantitative evaluation of TNF-α (left) and IL-1β (right) in the ischemic hemispheres. GAS inhibited the expression of TNF-α (F 4,20 = 31.66, P < 0.01) and IL-1β (F 4,20 = 19.03, P < 0.01) in the ischemic hemispheres. *P < 0.05 vs. MCAO + vehicle group
Discussion
Ischemic pathogenesis is rapidly initiated within minutes after the onset of MCAO, which involved a series of subsequent biochemical events including oxidative stress, inflammatory responses, programmed cell death, etc. [3, 34]. These events eventually lead to irreversible damage of proteins, nucleic acids and cell organelles that cause cellular dysfunction and neuronal death. The structure of GAS is a phenol 4-Hydroxybenzyl alcohol 4-O-beta-D-glucopyranoside (C13H18O7) and its molecular weight is 286.28. It was found that GAS could bind with human fibrinogen at the atomic level [35], and the entry of GAS into the brain was rapid via the femoral vein administration [36]; Additionally, GAS could penetrate through the blood–brain barrier into brain [37], we believe that it could allow for peritoneal injection. Previous study indicated that pretreatment of GAS could decrease the glutamate/GABA ratio during ischemia and reperfusion [38], and it could markedly decreased the infarct volume and edema volume in MCAO rats at 24 h after reperfusion [28, 39]. But whether this effect of GAS in neuroprotection is transient or permanent is still unknown. Also the mechanism was not identified yet. As described in the preciously studies, the definite infarct and iNOS expression in microglia were not detected until 24 h after reperfusion [40], and the long lasting neuroprotective effect could be observed 7 days after reperfusion [41, 42]. In the present study, we showed that GAS reduced the infarct volume ratio from ischemia-induced injury when administered one hour after MCAO. Besides, GAS treatment daily for 7 consecutive days showed long lasting neuroprotective effects even at 7 days after reperfusion. Since cerebral ischemia can induced both necrotic and apoptotic cell death [43], the present study investigated the cerebral damage in the primary somatosensory cortex and CA1 of hippocampus, which were related to the sensorimotor and memory deficits in the brain ischemia [44–46], and the expression of cleaved Caspase 3, Bax and Bcl-2 in the ischemic hemispheres as well. Caspase-3 can enzymatically digest specific substrates and inhibit DNA repair enzymes, thus leading to breakage of the chromosome into small fragments and eventual apoptosis. Moreover, proteins in the Bcl-2 family are central to the intrinsic apoptosis pathway, it is known that the pro-apoptotic protein Bax and the anti-apoptotic protein Bcl-2 can migrate from the cytoplasm to mitochondria, which are distributed in a manner that is consistent with mitochondrial release of cytochrome C and caspase [47]. The results showed that GAS treatment presented a significant functional recovery and morphology change and preserved the expression of Bcl-2 protein and suppressed expression of cleaved Caspase 3 and Bax induced by cerebral ischemia at 24 h and 7 days after reperfusion. These data provide direct evidence that GAS has a significant neuroprotective effect in cerebral ischemia.
We chose a transient cerebral ischemia model (1 h MCAO) and didn’t test whether GAS has a neuroprotective effect against permanent cerebral ischemic injury in the current study, mainly because GAS was reported to have an anti-oxidative and anti-inflammation property, which are two of the major characteristics for ischemic-reperfusion injury in the brain. It has been reported that several neuroprotective agents or strategies like dextromethorphan, riluzole, pioglitazone or HBO preconditioning are selectively effective only in transient ischemic injury models but not permanent ischemic injury models [48–50]. What’s more, the basic pathophysiological processes are in ways different between transient and permanent cerebral ischemia including the pattern of neurotoxic excitatory amino acid-glutamate release [51]. And the inflammatory response were more intense in permanent ischemia models as described in previous report that leukocyte infiltration was elevated at 24 h after reperfusion but did not further increase in transient ischemia model while permanent ischemia caused much more infiltration T cells in the brain at 5 days after ischemia [52]. In the current study, GAS significantly inhibited inflammatory cytokine expression in the ischemic brain within 24 h after ischemia, which effectively inhibited the acute inflammatory response in transient cerebral ischemia and provided effective neuroprotection. As a potent anti-inflammatory agent, repeated injection of GAS after cerebral ischemia might provide neuroprotective effect for permanent cerebral ischemic injury in animal models. But this effect and optimal dosage of GAS administration needs to be explored.
The phosphatidylinositol 3-kinase (PI3-K)/Akt pathway has been shown to play critical roles in the regulation of cell growth, proliferation, differentiation, survival, and intracellular trafficking. In addition, this pathway negatively regulates LPS-induced acute inflammatory responses [53, 54]. Furthermore, PI3-K/Akt signaling was required for the activation of nuclear factor-erythroid 2-related factor 2 (Nrf2) [55], which is a major regulator for several cytoprotective factors such as anti-oxidative enzymes, anti-inflammatory factors and transcriptional factors [56, 57]. Activation of the Nrf2 plays a pivotal role in enhancing the endogenous defense mechanism by which the brain protects itself against progressive ischemic damage and recovers from stroke [58, 59]. Moreover, the activation of Nrf2 can coordinately up-regulate the expression of several anti-oxidative enzymes such as heme oxygenase-1 (HO-1) and superoxide dismutase1 (SOD1) [60, 61]. All of them were recognized to play important roles in combating oxidative stress. Therefore, therapies targeting the Akt/Nrf2 pathway have provided a promising target for stroke treatment [62]. Previous studies showed that GAS could inhibit NF-κB activation, MAPKs phosphorylation and pro-inflammatory mediators and pro-inflammatory cytokines production by activating PI3-K/Akt pathway [63]. GAS could also up-regulate gene expression of Nrf2 to protect primary cultured rat hippocampal neurons against amyloid-beta peptide-induced neurotoxicity [27]. In addition, Studies have revealed a therapeutic window of approximately 6 h between the onset of ischemia and irreversible neuronal death [64, 65]. Therefore, it is desirable that neuroprotective interventions should be attempted before stroke occurs or very soon afterward.
To further elucidate the mechanism of GAS on alleviating MCAO-induced brain injury, the present study investigated the effects of GAS on the amount of SOD and MDA production, which is one of the classic oxidative stress markers to directly reflect the rate and extent of lipid peroxidation. In addition, the protein expression of phosphorylation level of Akt and Nrf2, as well as anti-oxidative enzymes (HO-1/SOD1) and pro-inflammatory cytokines (TNF-α/IL-1β) to reveal the role of Akt and Nrf2 signaling in the anti-oxidative and anti-inflammatory effect of GAS after cerebral ischemic injury at 6 h after reperfusion, which is based on the therapeutic window that there are approximately 6 h between the onset of ischemia and irreversible neuronal death [64, 65]. Our findings showed that MDA production was decreased after GAS treatment, as well as the expression of TNF-α and IL-1β. However, the production and expression of HO-1 and SOD1 in the ischemic brain was increased by GAS treatment. Furthermore, GAS significantly increased the phosphorylation Akt and Nrf2 expression. These results suggest that GAS regulates Akt phosphorylation and Nrf2 expression, thereby enhancing the protective defense mechanisms through anti-oxidative and anti-inflammatory pathway.
In conclusion, our findings suggest that GAS had neuroprotective effects against cerebral ischemic injury in a mice MCAO model. Notably, we also found that neuroprotective effect of GAS was associated with the preservation of anti-oxidative enzymes and inhibition of pro-inflammatory cytokines. This effect of GAS may be intermediated by the activation of Akt/Nrf2 pathway. These findings underscore GAS as a potential therapy for the treatment of ischemic brain injury, and it would be a useful agent when co-application to other drugs (such as tPA). However, the involvement of Akt/Nrf2 in the neuroprotective effects of GAS and the mechanism of how GAS affects the phosphorylation status of Akt and the expression of Nrf2 needs to be further investigated. Further studies are needed to explore the detailed neurobiological and cellular mechanisms underlying GAS treatment.
Abbreviations
- GAS:
-
Gastrodin
- Nrf2:
-
Nuclear factor erythroid 2-related factor 2
- TNF-α:
-
Tumor necrosis factor-α
- IL-1β:
-
Interleukin-1β
- ROS:
-
Reactive oxygen species
- CAT:
-
Catalase
- SOD:
-
Superoxide dismutase
- MCAO:
-
Middle cerebral artery occlusion
- PI3-K:
-
Phosphatidylinositol 3-kinase
- HO-1:
-
Heme oxygenase-1
- MDA:
-
Malondialdehyde
References
Russo T, Felzani G, Marini C (2011) Stroke in the very old: a systematic review of studies on incidence, outcome, and resource use. J Aging Res 2011:108785
Donnan GA, Fisher M, Macleod M, Davis SM (2008) Stroke. Lancet 371:1612–1623
Lakhan SE, Kirchgessner A, Hofer M (2009) Inflammatory mechanisms in ischemic stroke: therapeutic approaches. J Transl Med 7:97
Hacke W, Kaste M, Bluhmki E, Brozman M, Davalos A, Guidetti D, Larrue V, Lees KR, Medeghri Z, Machnig T, Schneider D, von Kummer R, Wahlgren N, Toni D (2008) Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med 359:1317–1329
Zhang B, Sun XJ, Ju CH (2011) Thrombolysis with alteplase 4.5–6 hours after acute ischemic stroke. Eur Neurol 65:170–174
Tan Z, Li X, Turner RC, Logsdon AF, Lucke-Wold B, DiPasquale K, Jeong SS, Chen R, Huber JD, Rosen CL (2014) Combination treatment of r-tPA and an optimized human apyrase reduces mortality rate and hemorrhagic transformation 6 h after ischemic stroke in aged female rats. Eur J Pharmacol 738:368–373
Jin X, Liu J, Liu W (2014) Early ischemic blood brain barrier damage: a potential indicator for hemorrhagic transformation following tissue plasminogen activator (tpa) thrombolysis? Curr Neurovasc Res 11(3):254-262
Chan PH (1996) Role of oxidants in ischemic brain damage. Stroke 27:1124–1129
Chen H, Yoshioka H, Kim GS, Jung JE, Okami N, Sakata H, Maier CM, Narasimhan P, Goeders CE, Chan PH (2011) Oxidative stress in ischemic brain damage: mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid Redox Signal 14:1505–1517
Jin R, Yang G, Li G (2010) Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol 87:779–789
Amantea D, Nappi G, Bernardi G, Bagetta G, Corasaniti MT (2009) Post-ischemic brain damage: pathophysiology and role of inflammatory mediators. FEBS J 276:13–26
Niizuma K, Endo H, Chan PH (2009) Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival. J Neurochem 109(Suppl 1):133–138
Deb P, Sharma S, Hassan KM (2010) Pathophysiologic mechanisms of acute ischemic stroke: an overview with emphasis on therapeutic significance beyond thrombolysis. Pathophysiology 17:197–218
Minami M, Katayama T, Satoh M (2006) Brain cytokines and chemokines: roles in ischemic injury and pain. J Pharmacol Sci 100:461–470
Qiao H, Zhang X, Zhu C, Dong L, Wang L, Xing Y, Wang C, Ji Y, Cao X (2012) Luteolin downregulates TLR4, TLR5, NF-kappaB and p-p38MAPK expression, upregulates the p-ERK expression, and protects rat brains against focal ischemia. Brain Res 1448:71–81
Du Y, Zhang X, Ji H, Liu H, Li S, Li L (2012) Probucol and atorvastatin in combination protect rat brains in MCAO model: upregulating Peroxiredoxin2, Foxo3a and Nrf2 expression. Neurosci Lett 509:110–115
Li M, Zhang X, Cui L, Yang R, Wang L, Liu L, Du W (2011) The neuroprotection of oxymatrine in cerebral ischemia/reperfusion is related to nuclear factor erythroid 2-related factor 2 (nrf2)-mediated antioxidant response: role of nrf2 and hemeoxygenase-1 expression. Biol Pharm Bull 34:595–601
Kim HJ, Moon KD, Lee DS, Lee SH (2003) Ethyl ether fraction of Gastrodia elata Blume protects amyloid beta peptide-induced cell death. J Ethnopharmacol 84:95–98
Kim HJ, Moon KD, Oh SY, Kim SP, Lee SR (2001) Ether fraction of methanol extracts of Gastrodia elata, a traditional medicinal herb, protects against kainic acid-induced neuronal damage in the mouse hippocampus. Neurosci Lett 314:65–68
Park S, da Kim S, Kang S (2011) Gastrodia elata Blume water extracts improve insulin resistance by decreasing body fat in diet-induced obese rats: vanillin and 4-hydroxybenzaldehyde are the bioactive candidates. Eur J Nutr 50:107–118
Xu X, Lu Y, Bie X (2007) Protective effects of gastrodin on hypoxia-induced toxicity in primary cultures of rat cortical neurons. Planta Med 73:650–654
Dai JN, Zong Y, Zhong LM, Li YM, Zhang W, Bian LG, Ai QL, Liu YD, Sun J, Lu D (2011) Gastrodin inhibits expression of inducible NO synthase, cyclooxygenase-2 and proinflammatory cytokines in cultured LPS-stimulated microglia via MAPK pathways. PLoS One 6:e21891
Hsieh MT, Wu CR, Chen CF (1997) Gastrodin and p-hydroxybenzyl alcohol facilitate memory consolidation and retrieval, but not acquisition, on the passive avoidance task in rats. J Ethnopharmacol 56:45–54
Peng Z, Wang H, Zhang R, Chen Y, Xue F, Nie H, Wu D, Wang Y, Tan Q (2013) Gastrodin ameliorates anxiety-like behaviors and inhibits IL-1beta level and p38 MAPK phosphorylation of hippocampus in the rat model of posttraumatic stress disorder. Physiol Res 62:537–545
Wang H, Zhang R, Qiao Y, Xue F, Nie H, Zhang Z, Wang Y, Peng Z, Tan Q (2014) Gastrodin ameliorates depression-like behaviors and up-regulates proliferation of hippocampal-derived neural stem cells in rats: involvement of its anti-inflammatory action. Behav Brain Res 266:153–160
Shu C, Chen C, Zhang DP, Guo H, Zhou H, Zong J, Bian Z, Dong X, Dai J, Zhang Y, Tang Q (2012) Gastrodin protects against cardiac hypertrophy and fibrosis. Mol Cell Biochem 359:9–16
Zhao X, Zou Y, Xu H, Fan L, Guo H, Li X, Li G, Zhang X, Dong M (2012) Gastrodin protect primary cultured rat hippocampal neurons against amyloid-beta peptide-induced neurotoxicity via ERK1/2-Nrf2 pathway. Brain Res 1482:13–21
Zeng X, Zhang S, Zhang L, Zhang K, Zheng X (2006) A study of the neuroprotective effect of the phenolic glucoside gastrodin during cerebral ischemia in vivo and in vitro. Planta Med 72:1359–1365
Zhang R, Peng Z, Wang H, Xue F, Chen Y, Wang Y, Tan Q (2014) Gastrodin ameliorates depressive-like behaviors and up-regulates the expression of BDNF in the hippocampus and hippocampal-derived astrocyte of rats. Neurochem Res 39:172–179
Wang Q, Peng Y, Chen S, Gou X, Hu B, Du J, Lu Y, Xiong L (2009) Pretreatment with electroacupuncture induces rapid tolerance to focal cerebral ischemia through regulation of endocannabinoid system. Stroke 40:2157–2164
Guo F, Jin WL, Li LY, Song WY, Wang HW, Gou XC, Mi YJ, Wang Q, Xiong L (2013) M9, a novel region of amino-Nogo-A, attenuates cerebral ischemic injury by inhibiting NADPH oxidase-derived superoxide production in mice. CNS Neurosci Ther 19:319–328
Tatlisumak T, Takano K, Carano RA, Miller LP, Foster AC, Fisher M (1998) Delayed treatment with an adenosine kinase inhibitor, GP683, attenuates infarct size in rats with temporary middle cerebral artery occlusion. Stroke 29:1952–1958
Li RC, Guo SZ, Lee SK, Gozal D (2010) Neuroglobin protects neurons against oxidative stress in global ischemia. J Cereb Blood Flow Metab 30:1874–1882
Choi YK, Cho GS, Hwang S, Kim BW, Lim JH, Lee JC, Kim HC, Kim WK, Kim YS (2010) Methyleugenol reduces cerebral ischemic injury by suppression of oxidative injury and inflammation. Free Radic Res 44:925–935
Liu Y, Tang X, Pei J, Zhang L, Liu F, Li K (2006) Gastrodin interaction with human fibrinogen: anticoagulant effects and binding studies. Chemistry 12:7807–7815
Wang Q, Chen G, Zeng S (2008) Distribution and metabolism of gastrodin in rat brain. J Pharm Biomed Anal 46:399–404
Lin LC, Chen YF, Lee WC, Wu YT, Tsai TH (2008) Pharmacokinetics of gastrodin and its metabolite p-hydroxybenzyl alcohol in rat blood, brain and bile by microdialysis coupled to LC-MS/MS. J Pharm Biomed Anal 48:909–917
Zeng X, Zhang Y, Zhang S, Zheng X (2007) A microdialysis study of effects of gastrodin on neurochemical changes in the ischemic/reperfused rat cerebral hippocampus. Biol Pharm Bull 30:801–804
Bie X, Chen Y, Han J, Dai H, Wan H, Zhao T (2007) Effects of gastrodin on amino acids after cerebral ischemia–reperfusion injury in rat striatum. Asia Pac J Clin Nutr 16(Suppl 1):305–308
Zhang N, Komine-Kobayashi M, Tanaka R, Liu M, Mizuno Y, Urabe T (2005) Edaravone reduces early accumulation of oxidative products and sequential inflammatory responses after transient focal ischemia in mice brain. Stroke 36:2220–2225
Liu H, Zhang X, Du Y, Ji H, Li S, Li L, Xing Y, Dong L, Wang C, Zhao K, Ji Y, Cao X (2012) Leonurine protects brain injury by increased activities of UCP4, SOD, CAT and Bcl-2, decreased levels of MDA and Bax, and ameliorated ultrastructure of mitochondria in experimental stroke. Brain Res 1474:73–81
Eady TN, Khoutorova L, Obenaus A, Mohd-Yusof A, Bazan NG, Belayev L (2014) Docosahexaenoic acid complexed to albumin provides neuroprotection after experimental stroke in aged rats. Neurobiol Dis 62:1–7
Broughton BR, Reutens DC, Sobey CG (2009) Apoptotic mechanisms after cerebral ischemia. Stroke 40:e331–e339
Borstad A, Schmalbrock P, Choi S, Nichols-Larsen DS (2012) Neural correlates supporting sensory discrimination after left hemisphere stroke. Brain Res 1460:78–87
Meehan SK, Dao E, Linsdell MA, Boyd LA (2011) Continuous theta burst stimulation over the contralesional sensory and motor cortex enhances motor learning post-stroke. Neurosci Lett 500:26–30
Kim MJ, Cho JH, Park JH, Ahn JH, Tae HJ, Cho GS, Yan BC, Hwang IK, Lee CH, Bae EJ, Won MH, Lee JC (2014) Impact of hyperthermia before and during ischemia-reperfusion on neuronal damage and gliosis in the gerbil hippocampus induced by transient cerebral ischemia. J Neurol Sci. doi:10.1016/j.jns.2014.11.015
Pape M, Engelhard K, Eberspacher E, Hollweck R, Kellermann K, Zintner S, Hutzler P, Werner C (2006) The long-term effect of sevoflurane on neuronal cell damage and expression of apoptotic factors after cerebral ischemia and reperfusion in rats. Anesth Analg 103:173–179
Britton P, Lu XC, Laskosky MS, Tortella FC (1997) Dextromethorphan protects against cerebral injury following transient, but not permanent, focal ischemia in rats. Life Sci 60:1729–1740
Xiong L, Zhu Z, Dong H, Hu W, Hou L, Chen S (2000) Hyperbaric oxygen preconditioning induces neuroprotection against ischemia in transient not permanent middle cerebral artery occlusion rat model. Chin Med J (Engl) 113:836–839
Shimazu T, Inoue I, Araki N, Asano Y, Sawada M, Furuya D, Nagoya H, Greenberg JH (2005) A peroxisome proliferator-activated receptor-gamma agonist reduces infarct size in transient but not in permanent ischemia. Stroke 36:353–359
Yang Y, Li Q, Miyashita H, Yang T, Shuaib A (2001) Different dynamic patterns of extracellular glutamate release in rat hippocampus after permanent or 30-min transient cerebral ischemia and histological correlation. Neuropathology 21:181–187
Zhou W, Liesz A, Bauer H, Sommer C, Lahrmann B, Valous N, Grabe N, Veltkamp R (2013) Postischemic brain infiltration of leukocyte subpopulations differs among murine permanent and transient focal cerebral ischemia models. Brain Pathol 23:34–44
Zong Y, Sun L, Liu B, Deng YS, Zhan D, Chen YL, He Y, Liu J, Zhang ZJ, Sun J, Lu D (2012) Resveratrol inhibits LPS-induced MAPKs activation via activation of the phosphatidylinositol 3-kinase pathway in murine RAW 264.7 macrophage cells. PLoS One 7:e44107
Zhang WJ, Wei H, Hagen T, Frei B (2007) Alpha-lipoic acid attenuates LPS-induced inflammatory responses by activating the phosphoinositide 3-kinase/Akt signaling pathway. Proc Natl Acad Sci U S A 104:4077–4082
Wang L, Chen Y, Sternberg P, Cai J (2008) Essential roles of the PI3 kinase/Akt pathway in regulating Nrf2-dependent antioxidant functions in the RPE. Invest Ophthalmol Vis Sci 49:1671–1678
Ren J, Fan C, Chen N, Huang J, Yang Q (2011) Resveratrol pretreatment attenuates cerebral ischemic injury by upregulating expression of transcription factor Nrf2 and HO-1 in rats. Neurochem Res 36:2352–2362
Fujita K, Maeda D, Xiao Q, Srinivasula SM (2011) Nrf2-mediated induction of p62 controls Toll-like receptor-4-driven aggresome-like induced structure formation and autophagic degradation. Proc Natl Acad Sci U S A 108:1427–1432
Wang B, Cao W, Biswal S, Dore S (2011) Carbon monoxide-activated Nrf2 pathway leads to protection against permanent focal cerebral ischemia. Stroke 42:2605–2610
Li L, Zhang X, Cui L, Wang L, Liu H, Ji H, Du Y (2013) Ursolic acid promotes the neuroprotection by activating Nrf2 pathway after cerebral ischemia in mice. Brain Res 1497:32–39
Cho BO, Ryu HW, Jin CH, Choi DS, Kang SY, Kim DS, Byun MW, Jeong IY (2011) Blackberry extract attenuates oxidative stress through up-regulation of Nrf2-dependent antioxidant enzymes in carbon tetrachloride-treated rats. J Agric Food Chem 59:11442–11448
Shah ZA, Li RC, Thimmulappa RK, Kensler TW, Yamamoto M, Biswal S, Dore S (2007) Role of reactive oxygen species in modulation of Nrf2 following ischemic reperfusion injury. Neuroscience 147:53–59
Wu J, Li Q, Wang X, Yu S, Li L, Wu X, Chen Y, Zhao J, Zhao Y (2013) Neuroprotection by curcumin in ischemic brain injury involves the Akt/Nrf2 pathway. PLoS One 8:e59843
Yang P, Han Y, Gui L, Sun J, Chen YL, Song R, Guo JZ, Xie YN, Lu D, Sun L (2013) Gastrodin attenuation of the inflammatory response in H9c2 cardiomyocytes involves inhibition of NF-kappaB and MAPKs activation via the phosphatidylinositol 3-kinase signaling. Biochem Pharmacol 85:1124–1133
Williams AJ, Berti R, Dave JR, Elliot PJ, Adams J, Tortella FC (2004) Delayed treatment of ischemia/reperfusion brain injury: extended therapeutic window with the proteosome inhibitor MLN519. Stroke 35:1186–1191
Xu Z, Croslan DR, Harris AE, Ford GD, Ford BD (2006) Extended therapeutic window and functional recovery after intraarterial administration of neuregulin-1 after focal ischemic stroke. J Cereb Blood Flow Metab 26:527–535
Acknowledgments
This work was funded by the National Natural Science Foundation of China (Grant No. 81401109, 81171285 and 81371478) and No. 13QNP28, Youth Training Project of the Health Department of General Logistics Department to Dr. Jiao Deng.
Conflict of interest
The authors declared that there are no conflicts of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Zhengwu Peng, Shiquan Wang and Guanjie Chen has contributed equally to this work.
Rights and permissions
About this article

Cite this article
Peng, Z., Wang, S., Chen, G. et al. Gastrodin Alleviates Cerebral Ischemic Damage in Mice by Improving Anti-oxidant and Anti-inflammation Activities and Inhibiting Apoptosis Pathway. Neurochem Res 40, 661–673 (2015). https://doi.org/10.1007/s11064-015-1513-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-015-1513-5