
Abstract
Amyloid-β (Aβ)-induced mitochondrial dysfunction has been recognized as a prominent, early event in Alzheimer’s disease (AD). Therefore, therapeutics targeted to improve mitochondrial function could be beneficial. Quercetin, a bioflavanoid, has been reported to have potent neuro-protective effects, but its preventive effects on Aβ-induced mitochondrial dysfunction and cognitive impairment have not been well characterised. Three-month-old APPswe/PS1dE9 transgenic mice were randomly assigned to a vehicle group, two quercetin (either 20 or 40 mg kg−1 day−1) groups, or an Aricept (2 mg kg−1 day−1) group. After 16 weeks of treatment, we observed beneficial effects of quercetin (40 mg kg−1 day−1), including lessening learning and memory deficits, reducing scattered senile plaques, and ameliorating mitochondrial dysfunction, as evidenced by restoration of mitochondrial membrane potential, reactive oxygen species and ATP levels in mitochondria isolated from the hippocampus compared to control. Furthermore, the AMP-activated protein kinase (AMPK) activity significantly increased in the quercetin-treated (40 mg kg−1 day−1) group. These findings suggest that a reduction in plaque burden and mitochondrial dysfunction through the activation of AMPK may be one of the mechanisms by which quercetin improves cognitive functioning in the APPswe/PS1dE9 transgenic mouse model of AD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a degenerative neurological disease that is clinically characterised by progressive cognitive dysfunction [1, 2]. Increasing evidence shows that mitochondrial dysfunction is a main cellular event in the pathogenesis and progression of AD [ 3, 4 ]. The progressive deposition of amyloid-β (Aβ) in the mitochondria, which is the most important pathogenic factor in AD, can trigger mitochondrial dysfunction [5] through the disruption of the mitochondrial membrane potential (MMP) [6, 7], elevation of reactive oxygen species (ROS) production and cellular metabolic deficiencies [8]. The standard treatment for symptomatic AD relies primarily on acetyl-cholinesterase (AChE) inhibitors, such as Aricept. However, AChE inhibition alleviates the symptoms of AD, but has only a modest beneficial effect on neurodegeneration [9]. Therapeutic applications targeted to improve mitochondrial function in AD are therefore very promising and could aid in improvement of both pathological and cognitive aspects of the disease.
Quercetin (3,5,7,30,40-pentahydroxyflavone, a bioflavonoid), frequently found in consumable foods including apples, berries, onions, tea and brassica vegetables, has many beneficial effects on human health, including cardiovascular protection, anticancer activity, cataract prevention, antiviral activity and anti-inflammatory effects [10]. It has been reported that quercetin can pass through the blood–brain barrier in situ models [11]. Quercetin may have neuro-protective effects and slow down the progression of degenerative diseases. Quercetin has been reported to attenuate behavioural alterations and cognitive impairment in Parkinson’s disease models [12] and chronic cerebral ischemia models [13]. Moreover, quercetin administration improves cognitive deficits in colchicines [14], scopolamine [15] and aluminium [16] induced learning and memory impairment models. Quercetin has been indicated to penetrate in and rigidify the bilayer of membranes and reduce the interaction of the membrane with the Aβ peptide (25–35) in vitro (Tedeschi et al. 2010). Furthermore, study shows that quercetin decreases amyloid precursor protein (APP) maturation through regulating APP processing [17] and prevents Aβ-induced neurotoxicity in primary neuronal cells [18]. To date, studies of quercetin on AD are mainly focused on its antioxidant ability and relationship to oxidative stress. The potential effects of quercetin on amyloid neuropathy and mitochondrial dysfunction have not been reported.
The APPswe/PS1dE9 transgenic mouse model of AD exhibits senile plaque formation and amyloid neuropathy [ 19 ], mitochondrial dysfunction and progressive behavioural deficits [20, 21]. Compared with the colchicines, scopolamine or aluminium induced animal models of Alzheimer’s disease, the APPswe/PS1dE9 transgenic mouse model might be better to mimics the progression of the pathology of human AD. This model can therefore be used to comprehensively evaluate the effect of quercetin on Aβ-induced neurotoxicity and mitochondrial dysfunction, which has not been studied yet. In this study, the effect of long-term quercetin consumption on the prevention of memory loss, Aβ-induced neurotoxicity and mitochondrial dysfunction were studied in the APPswe/PS1dE9 transgenic mouse model of Alzheimer’s disease.
Experimental Procedures
Animals
APPswe/PS1dE9 mice (C57/BL) used in this study was generated as previously described [22, 23]. Mice express a mouse–human hybrid transgene containing the extracellular and intracellular regions of the mouse sequence and a human sequence within the Aβ domain with Swedish mutations (K594N/M595L), and express the human presenile-1 deleted exon-9. Protocols were conducted according to the University Policies on the Use and Care of Animals and were approved by the Institutional Animal Experiment Committee of Henan University of Science and Technology, China.
Group and Treatment
Three-month-old APPswe/PS1dE9 transgenic mice were randomly assigned among treatment groups. APPswe/PS1dE9 transgenic mice were fed either AIN-76A chow (Dyets Inc., Bethlehem, PA, USA) containing a low dose of quercetin (20 mg kg−1 day−1; n = 12), a high dose of quercetin (40 mg kg−1 day−1; n = 12), and a dose of Aricept (2 mg kg−1 day−1; n = 12) or no drug (n = 12) for 16 weeks before being killed. Non-transgenic littermates were fed chow as wild type (n = 12). Each group included six males and six females. The dose of quercetin was selected based on other experimental studies [24, 25]. Aricept, an inhibitor of acetyl-cholinesterase, is presently used in long-term treatments for patients with AD. The administered dose of Aricept was calculated from the weight of the mice to be equivalent to the human dose.
Behavioural Tests
Novel Object Recognition Test
The test procedure consisted of three sessions: habituation, training, and retention. Each mouse was habituated to the box (30 × 30 × 35 cm), with 10 min of exploration in the absence of objects for 3 days (habituation session). During the training session, two objects were placed at the back corner of the box. A mouse was then placed in the box and the total time spent exploring the two objects was recorded for 10 min. During the retention session, the mice were placed back in the same box 24 h after the training session, in which one of the familiar objects used during the training was replaced with a novel object. The animals were then allowed to explore freely for 5 min, the exploration time for the familiar (TF) or the new object (TN) during the test phase was recorded. A recognition index, a ratio of the amount of time spent exploring any one of the two objects (training session) or the novel object (retention session) over the total time spent exploring both objects, was used to measure cognitive function. To control for odor cues, the OF arena and the objects were thoroughly cleaned with 10 % odorless soap, dried, and ventilated for a few minutes between mice [26, 27].
Morris Water Maze
Spatial learning and memory was tested using the Morris water maze, performed 1 day after the end of the open-field test. The protocol for the Morris water maze test was modified from previously reported methods [28, 29]. Briefly, the apparatus included a pool with a diameter of 100 cm that was filled with opaque water at approximately 22 ± 1 °C. An escape platform (15 cm in diameter) was placed 0.5 cm below the water surface. Geometric objects with contrasting colours were set at the remote ends of the water tank as references. Room temperature was constant, and the lighting was even throughout the room. Spatial memory is assessed by recording the latency time for the animal to escape from the water onto a submerged escape platform during the learning phase. The mice were subjected to four trials per day for five consecutive days. The mice were allowed to stay on the platform for 15 s before and after each trial. The time that it took for an animal to reach the platform (latency period) was recorded. Twenty-four hours after the learning phase, the mice swam freely in the water tank without the platform for 60 s, and the time spent in the region, and number of passes through the region and the quadrant of the original platform were recorded. Monitoring was performed with a video tracking system (Noldus Ltd, Ethovision XT, Holland).
Tissue Preparation
The mice (n = 6) following the behavioural test were randomly chosen and deeply anesthetized with sodium pentobarbital (100 mg kg−1 intraperitoneally). Brain were removed and dissected through the midsagittal plane. One hemisphere was fixed in formalin and embedded in paraffin. For each specimen, 30 serial sections of 5 μm thickness were coronally sliced for two to three such series, spaced 50 μm apart. The remaining hemi brain were were directly homogenised in RIPA buffer containing 0.1 % PMSF and 0.1 % protease inhibitor cocktail (Sigma, MO, USA). The lysates were centrifuged at 14,000g for 30 min at 4 °C and the supernatant was used for protein analyses. The protein concentration in supernatants was determined using the BCA method.
Histological Analysis
To demonstrate fibrillar Aβ deposition, Thioflavine-S staining was used, which is commonly used for fluorescent staining of plaques [30, 31]. Brain tissue from mice (n = 6) following behavioural tests was fixed in 10 % neutral buffered formalin and mounted in paraffin blocks. After deparaffinization and hydration, the sections were washed in PBS and incubated in 0.25 % potassium permanganate and 1 % oxalic acid until they appeared white. The sections were then washed in water and stained for 3 min with a solution of 0.015 % Thioflavin-S in 50 % ethanol. Finally, the sections were washed in 50 % ethanol and in water, then dried, and dipped in Histo-Clear before being cover-slipped with Permount [32]. Sections stained with Thioflavin-S were visualized and images were captured by microscope with a digital camera attached to a computer (Nikon, Melville, NY). Images were analyzed using Aperio’s ImageScope Viewer software (Aperio. Technologies).
Preparation of Isolated Mitochondria
After behavioural tests, the mice were euthanatized using CO2 asphyxiation and brain mitochondria were isolated, performed using a standard procedure [33]. First, brains were quickly removed and placed on ice. Brain regions of interest were carefully dissected following anatomical guidelines and placed in a glass Dounce homogenizer containing five times the volume of isolation buffer [215 mM mannitol, 75 mM sucrose, 0.1 % BSA, 1 mM EGTA, 20 mM HEPES (Na+), pH 7.2]. Following homogenisation, a low-speed spin (1,300g for 5 min) to remove unbroken cells and nuclei was performed. The supernatant was carefully placed in fresh tubes, topped off with isolation buffer and spun down again at 13,000g for 10 min. The supernatant was discarded and the resultant mitochondrial pellets were suspended in 500 μl of isolation buffer with 1 mM EGTA (215 mM mannitol, 75 mM sucrose, 0.1 % BSA, 20 mM HEPES (Na+), pH 7.2) and 0.1 % digitonin (in DMSO) was added to the pellets to disrupt the synaptosomes. After 5 min, samples were brought to a final volume of 2 ml using isolation buffer containing 1 mM EGTA and centrifuged at 13,000g for 15 min. Next the pellets were resuspended in isolation buffer without EGTA (75 mM sucrose, 215 mM mannitol, 0.1 % BSA and 20 mM HEPES with the pH adjusted to 7.2 using KOH) and was centrifuged at 10,000g for 10 min. The final mitochondrial pellet was suspended in isolation buffer without EGTA to yield a final protein concentration of approximately 10 mg/ml and immediately stored on ice. To normalise the results, the protein concentrations were determined with all the samples on the same micro well plate using a BCA protein assay kit. For all mitochondrial analyses, the brain areas of interest from 3 mice of the same genotype were combined to form a single homogenate that was then assayed in triplicate. We performed the experiment twice (n = 2) with 6 quercetin treated APPswe/PS1dE9 mice, six control APPswe/PS1dE9 mice and 6 nontransgenic mice.
Membrane Potential Measurements on Isolated Mitochondria
A 200 μM stock solution of JC-1 (5,5,6,6-tetrachloro-1,1,3,3-tetraethylbenzimidazolylcarbocyanine iodide) was made using DMSO as the solvent. The assay buffer contained mitochondrial isolation buffer with the addition of 5 mM pyruvate and 5 mM malate. 150 μl of assay buffer and 20 μl (1.2 mg/ml final concentration) of mitochondria were added to the wells of a 96 well black, clear bottom microplate (Corning) followed by the addition of 1 μM JC-1 and mixed gently. The microplate was covered with aluminium foil and left at room temperature for 20 min before reading. Fluorescence (excitation 530/25 nm, emission 590/35 nm) was then measured.
Reactive Oxygen Species Production from Isolated Mitochondria
Mitochondrial ROS production was measured following incubation of isolated mitochondria with 25 μM 2,7-dichlorodihydrofluorescein diacetate for 20 min and then the DCF fluorescence (excitation filter 485/20 nm, emission filter 528/20 nm) was read as previously described [34]. In short, 100 μg (0.8 mg/ml final concentration) of isolated mitochondria were added to 120 μl of Kcl-based respiration buffer with 5 mM pyruvate and 2.5 mM malate added as respiratory substrates and 25 μM 2,7-dichlorodihydrofluorescein diacetate. Mitochondrial ROS production in the presence of oligomycin (to increase ROS production) or FCCP (to decrease ROS production) were performed to ensure measurement values were within the range of the indicator.
ATP Level Determination in Isolated Mitochondria
ATP was determined luminometrically using ATP bioluminescence assay kit (Sigma, St. Louis, MO, USA) according to the provided protocol. Mitochondrial samples were assayed for ATP content using the ATP dependence of the light emitting luciferase-catalyzed oxidation of luciferin. ATP concentration was calculated according to a standard curve and related to protein content assessed using the method of Lowry et al. (1951).
Western Blot Analysis
Equal amounts of soluble protein were separated by SDS-PAGE and transferred onto a nitrocellulose membrane (Immobilon NC; Millipore, Molsheim, France). Immunoblotting was carried out with antibodies specific for p-AMP-activated protein kinase (AMPK) 172 (1:1,000), AMPK (1:1,000, Cell Signalling Technology). Primary antibodies were visualised with anti-rabbit HRP conjugated secondary antibodies (Santa Cruz Biotechnology, Inc.) using a chemiluminescent detection system (Western blotting Luminal Reagent; Santa Cruz Biotechnology, Inc.). Variations in sample loading were corrected by normalising to GAPDH levels.
Statistical Analysis
All data were expressed as the mean ± SEM. For the Morris water maze tests, escape latency in the hidden platform trial were analysed with two-way ANOVA of repeated measures, while one-way ANOVA was conducted on the data obtained from the probe trial. The recognition index in the novel object recognition test was analysed by one-way ANOVA, followed by LSD (equal variances assumed) or Dennett’s (equal variances not assumed) for a post hoc test between groups. Histological assays and mitochondrial data were analysed by one-way ANOVA, followed by LSD. All analyses were performed with SPSS statistical package (version 13.0 for Windows, SPSS Inc., USA). Differences were considered significant at a p value <0.05.
Results
Behavioural Test
Quercetin Ameliorates Recognition Memory of APPswe/PS1dE9 Mice in Novel Object Recognition
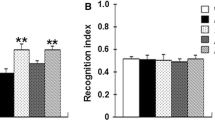
To evaluate cognitive function, a novel object recognition test was carried out in WT, controls APPswe/PS1dE9 mice and treated APPswe/PS1dE9 mice after 16 weeks of drug administration. In the test section, there was a significant overall group difference in the recognition index [F (4, 55) = 8.80, p < 0.01] among the five groups. Compared with WT mice, the recognition index (p < 0.01) was significantly reduced in APPswe/PS1dE9 mice. Mice treated with 40 mg kg−1 quercetin markedly increased the recognition index by 56.4 % (Fig. 1a). The 20 mg kg−1 quercetin group showed increase of the recognition index. However, this increase was not statistically significant. In addition, there was no significant difference in the recognition index (Fig. 1b) in training session between the five groups of mice (p > 0.05).

Effect of quercetin on the recognition memory in APPswe/PS1dE9 transgenic mice detected by a novel object recognition test. Non-transgenic littermates (WT) were given a standard diet and were used as controls for experiments involving APPswe/PS1dE9 mice. APPswe/PS1dE9 mice were given a standard diet and were used as a vehicle control (APPswe/PS1dE9). Other groups of APPswe/PS1dE9 mice were treated with quercetin at a dose of 20 mg kg−1 day−1 (Quercetin20) or 40 mg kg−1 day−1 (Quercetin 40) for 16 weeks. Some APPswe/PS1dE9 mice were treated with Aricept at a dosage of 2 mg kg−1 day−1 in the same manner as those treated with quercetin and these mice were used as positive controls (Aricept). The recognition index in the test section a and training section b were measured. Values are presented as mean ± SEM. The analysis was performed using one-way ANOVA with a LSD post hoc test between groups. (n = 12, **p < 0.01 compared with APPswe/PS1dE9 mice)
Quercetin Improves the Learning and Memory of APPswe/PS1dE9 Mice in the Morris Water Maze
To assess spatial reference learning and memory function, all mice underwent testing in the Morris water maze after 16 weeks of drug administration. Spatial learning was assessed in the hidden platform task in all mice. As shown in Fig. 2a, there was a significant overall group difference in escape latency among the five groups [group effect: F (4, 55) = 11.38, p < 0.01; training day effect: F (4, 220) = 104.92, p < 0.01; group × training day interaction: F (16, 220) = 0.58, p > 0.05]. The latency to finding the submerged platform decreased every day, but the escape latency in APPswe/PS1dE9 mice was significantly longer than that of the WT group (p < 0.01). Quercetin-treated APPswe/PS1dE9 mice showed decreased escape latency compared with APPswe/PS1dE9 control mice, especially in the 40 mg kg−1 quercetin group (p < 0.01). The 20 mg kg−1 quercetin group showed a shortening of the escape latency. However, this decrease was not statistically significant (Fig. 2a).

Effect of quercetin on learning and memory in APPswe/PS1dE9 transgenic mice using the Morris water maze. Escape latency during 5 days of hidden platform tests a, the number of crossing the platform b, swimming speed c and path length d in the probe test were tabulated. All data are presented as mean ± SEM. (n = 12, **p < 0.01 compared with APPswe/PS1dE9 mice)
In the probe test, the frequency of crossing the platform was measured for 60 s on the 6th day after the last acquisition test. As shown in Fig. 2b, there was a significant overall group difference in the frequency of crossing the platform among the five groups [F (4, 55) = 8.10, p < 0.01]. The frequency was decreased by 61.93 % in APPswe/PS1dE9 mice compared with WT mice (p < 0.01). Compared with APPswe/PS1dE9 mice, the number of platform crossings significantly was increased by 1.41-fold in the Aricept-treated group (p < 0.01) and by 1.53-fold in the 40 mg kg−1 quercetin groups (p < 0.01) (Fig. 2b). In addition, there was no significant difference in swimming speed (Fig. 2c) and path length (Fig. 2d) in the probe test between the five groups of mice (p > 0.05).
Sex has been reported to modulate AD susceptibility or the course of the disease. However, Gender effects on the frequencies of Alzheimer’s disease increase with aging [35]. In this present study, for seven-month-old mice, there was no significant difference in the frequency of crossing the platform in the probe test between male mice and female mice among the five groups (p > 0.05).
Quercetin Treatment Reduces Plaque Pathology in APPswe/PS1dE9 Mice
Quercetin has been suggested to reduce Aβ-induced neurotoxicity by changing the properties of the bilayer structure [36] and regulating APP processing in vitro. To investigate whether quercetin has an effect on amyloid plaque formation in vivo, mice treated or not treated with quercetin were sacrificed, and their hippocampi of mice were stained with Thioavine-S, which specifically binds amyloid plaques. Compared with APPswe/PS1dE9 transgenic mice, quercetin-treated APPswe/PS1dE9 mice exhibited 38.5, 43.8 % fewer Thioavine-S positive compact plaques (p < 0.05), 37.4 and 41.5 % less plaque area (p < 0.05) in hippocampus and cortex. By contrast, Aricept treatment did not inhibit plaque formation (Fig. 3).

Quercetin treatment reduced plaque pathology in APPswe/PS1dE9 mice. Brain tissue from WT, APPswe/PS1dE9 mice, Quercetin 40-treated mice and Aricept-treated mice were utilised in standard pathological procedures, and sections were stained with Thioflavin-S to visualise the deposition of Aβ. a Representative brain sections showing that quercetin decreased Thioflavin-S immunoreactivity (scale bars, 1 cm). b Graphs showing plaque count and the percentage of area occupied by plaques in the cortex and hippocampus. All data are presented as mean ± SEM. (n = 6, *p < 0.05 compared with APPswe/PS1dE9 mice)
Quercetin Alleviates Mitochondrial Dysfunction in an AD Model
Decreased MMP, impaired ATP syntheses and enhanced ROS production are usually a direct consequence of mitochondrial dysfunction and might contribute additionally to mitochondrial damage that accumulates with age. The APPswe/PS1dE9 mice have obvious mitochondrial defects, exhibiting a 75 % decrease in MMP (p < 0.01), a 55.6 % decrease in ATP contents (p < 0.01) and a 2.21-fold increase in ROS production (p < 0.01) of hippocampal mitochondria when compared with WT control mice. Compared with control APPswe/PS1dE9 mice, treatment with quercetin (40 mg kg−1 day−1) can significantly attenuate mitochondrial damage as exhibited by in increasing MMP, ATP levels and decreasing ROS production by 2.0-fold, 87.5 and 65,6 %, respectively, in hippocampal mitochondria of APPswe/PS1dE9 mice. In mitochondria from the cortex, quercetin administration restored MMP, ATP levels and ROS production by 70–80 %. Quercetin similarly restored ATP levels by 72 %, but the MMP and ROS production in this brain region were slightly less affected, only being restored by 50–60 %. Aricept did not rescue the mitochondrial defects of APPswe/PS1dE9 mice.
Quercetin Increases AMPK Activity in an AD Model
The AMPK is a proline-directed ser/thr kinase that associates with Aβ and mitochondrial function in AD. As phosphorylated AMPK Thr172 (pAMPK172) is a marker of AMPK activation; total AMPK and pAMPK172 were measured by Western blot to evaluate the activation of AMPK. Quercetin treatment increased the pAMPK172 level by 53.1 % compared with control APPswe/PS1dE9 mice. In contrast, Aricept treatment did not obviously affect the activity of AMPK (Fig. 7a, b).
Discussion
In the present study, we found quercetin ameliorated cognitive deficits, as indicated by enhancing learning and memory (i.e., increasing recognition index by 56.4 % in the novel object recognition test and inducing a 1.53-fold increase in the crossing-target number in the probe test) in this animal model of AD. The results demonstrated that quercetin reduced senile plaques, ameliorated mitochondrial dysfunction as indicated by restoring MMP, ROS production and ATP levels by 50–85 % in mitochondria isolated from the hippocampus, cortex and striatum, and increased the activity of AMPK in the APPswe/PS1dE9 transgenic mouse model of AD.
Mitochondria have been proposed to act as central organelles in the regulation of aging and age related neurodegeneration because they control cellular energy status, ROS production, and apoptosis, all of which are important in determining lifespan [37]. Mitochondria constitute the major source of superoxide and other ROS within most tissues, generating approximately 85–90 % of total cellular superoxide [38]. Mitochondrial electron transport in aged tissues is less efficient, which impairs ATP synthesis and results in increased oxidant production [39]. The steady-state levels of oxidatively damaged molecules are dependent both on net ROS formation and clearance of damaged molecules [40]. Antioxidant defences also decline with age [41], making mitochondria even more vulnerable to oxidative injury. The resultant mitochondrial decay may eventually cause inadequate energy production and/or the loss of mitochondrial and cellular calcium homeostasis. Such changes could result in cellular apoptosis and also lead to the loss of cognitive function accompanying brain aging [42]. Previous studies demonstrated that Aβ can induce the impairment of oxidative phosphorylation [43–45], increase the ROS production [46], disturb membranes properties and alteration of mitochondrial dynamics [47]. Consequently, this triggered mitochondrial dysfunction and contributed to the development and progression of AD [46, 48–50]. Consistent with this, our experiments also demonstrated (in APPswe/PS1dE9 transgenic mice) impaired mitochondrial function (as indicated by a decrease in MMP, ATP levels and an increase in ROS production) and cognitive deficits. Increasing evidence indicates that mitochondrial dysfunction and increased oxidative stress occur as early events in cognitive aging and AD pathogenesis prior to the appearance of amyloid deposition. Mitochondrial fragmentation and reduced neurite outgrowth caused by Aβ can both be suppressed by mitochondrial targeted antioxidants, such as MitoQ and the cell-permeable SS31 peptide [51]. Therefore, agents that attenuate mitochondrial dysfunction, and provide neuroprotective benefits represent a promising strategy for AD treatment.
AMP-activated protein kinase, a key cellular regulator of energy metabolism [52], has been implicated in the regulation of mitochondrial function. Damaged mitochondria are the major sources of ROS in cells and are related to neurodegenerative diseases, such as AD. Activated AMPK can maintain intracellular redox status and decrease intracellular ROS by inhibiting NADPH oxidases activity [53] or by increasing the expression of antioxidant enzymes such as superoxide dismutase-2 [54] and uncoupling protein-2 [55, 56]. Moreover, a line of agents has been shown to inhibit radical induced stress through AMPK activation as well as induction of antioxidant enzymes [57, 58]. Activation of AMPK, or overexpression of constitutively activated AMPK, can enhance mitochondrial biogenesis and improve mitochondrial function in cell lines [59–61] and animal models [62, 63]. Conversely, the inhibition of AMPK leads to an increase in ROS production, a decrease in mitochondrial biogenesis and thus worsens mitochondrial dysfunction [64, 65]. Our results indicate that quercetin can significantly ameliorate mitochondrial dysfunction that is observed in APPswe/PS1dE9 transgenic mice to that which is observed in WT mice (Figs. 4, 5, 6). Quercetin also increased the activity of AMPK in vivo (Fig. 7). We speculate that quercetin ameliorates mitochondrial dysfunction and increases oxidative defense mechanisms by activating of AMPK. The reduction of mitochondrial dysfunction is an effective way of preventing cognitive deficits and attenuating neuronal atrophy [66, 67] that is observed in AD. The positive effect of quercetin on cognition might function by attenuating mitochondrial dysfunction through the activation of AMPK.

Analysis of isolated hippocampal mitochondrial function from APPswe/PS1dE9 mice treated with quercetin. MMP a, ROS Production b, and ATP levels c were examined. All data are presented as mean ± SEM (n = 6, **p < 0.01 compared with APPswe/PS1dE9 mice)

Analysis of isolated cortex mitochondrial function from APPswe/PS1dE9 mice treated with quercetin. MMP a, ROS production b, and ATP levels (c) were examined. All data are presented as mean ± SEM. (n = 6, **p < 0.01 compared with APPswe/PS1dE9 mice)

Analysis of isolated striatum mitochondrial function from APPswe/PS1dE9 mice treated with quercetin. MMP a, ROS production b, and ATP levels c were examined. All data are presented as mean ± SEM (n = 6, *p < 0.05, **p < 0.01 compared with APPswe/PS1dE9 mice)

Quercetin increases AMPK activity in vivo. Protein lysates were prepared from the brains of WT mice, APPswe/PS1dE9 mice, Aricept-treated mice, Quercetin 20-treated mice and Quercetin 40-treated mice. The abundance of phosphorylated AMPKα and the expression of total AMPK were determined by Western blotting a. The average blot densitometry of three independent experiments is shown b (n = 4, *p < 0.05 compared with APPswe/PS1dE9 mice)
Our results also indicate that quercetin prevented the formation of senile plaques by 26.4 % and reduced plaque area by 32.9 % in the hippocampus compared with the control APPswe/PS1dE9 mice (Fig. 4). The inhibition of AMPK promotes Aβ generation [68]. Conversely, the activation of AMPK can decrease Aβ deposition through regulating APP processing, APP distribution and promoting the clearance of intracellular Aβ in both animal models [69, 70] and cell lines [71–73], and attenuate Aβ-induced neurotoxicity [74]. Furthermore, quercetin can penetrate and rigidify the membrane bilayer and reduces the interaction of the membrane with the Aβ (25–35) peptide in vitro (Tedeschi et al. 2010). Quercetin ability to prevent the formation of senile plaques may be caused by an activation of AMPK or/and a rigidification of the cellular membrane of neurons. However, this hypothesis needs to be investigated further.
In conclusion, quercetin (40 mg kg−1) is a bioflavonoid found to reduce scattered senile plaques, alleviate Aβ-induced mitochondrial dysfunction, and improve cognitive impairment in APPswe/PS1dE9 transgenic mice. The regulation of AMPK activity may be the primary mechanism by which quercetin affects AD phenotypes. Quercetin shows therapeutic promise in an AD transgenic mouse model, and it may have potential as an AD therapeutic agent.
Abbreviations
- AD:
-
Alzheimer’s disease
- Aβ:
-
Amyloid-β
- MMP:
-
Mitochondrial membrane potential
- ROS:
-
Reactive oxygen species
- AMPK:
-
AMP-activated protein kinase
- APP:
-
Amyloid precursor protein
References
Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS (2005) Axonopathy and transport deficits early in the pathogenesis of Alzheimer;s disease. Science 307:1282–1288
Mattson MP (2004) Pathways towards and away from Alzheimer’s disease. Nature 430:631–639
Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443:787–795
Moreira PI, Cardoso SM, Santos MS, Oliveira CR (2006) The key role of mitochondria in Alzheimer’s disease. J Alzheimers Dis 9:101–110
Cha MY, Han SH, Son SM, Hong HS, Choi YJ, Byun J, Mook-Jung I (2012) Mitochondria-specific accumulation of amyloid beta induces mitochondrial dysfunction leading to apoptotic cell death. PLoS One 7:e34929
Moreira PI, Santos MS, Moreno A, Rego AC, Oliveira C (2002) Effect of amyloid beta-peptide on permeability transition pore: a comparative study. J Neurosci Res 69:257–267
Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD (2008) Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med 14:1097–1105
Beal MF (2005) Mitochondria take centre stage in aging and neurodegeneration. Ann Neurol 58:495–505
Martinez-Coria H, Green KN, Billings LM, Kitazawa M, Albrecht M, Rammes G, Parsons CG, Gupta S, Banerjee P, LaFerla FM (2010) Memantine improves cognition and reduces Alzheimer’s-like neuropathology in transgenic mice. Am J Pathol 176:870–880
Nagata H, Takekoshi S, Takagi T, Honma T, Watanabe K (1999) Antioxidative action of flavonoids, quercetin and catechin, mediated by the activation of glutathione peroxidase. Tokai J Exp Clin Med 24:1–11
Youdim KA, Qaiser MZ, Begley DJ, Rice-Evans CA, Abbott NJ (2004) Flavonoid permeability across an in situ model of the blood–brain barrier. Free Radic Biol Med 36:592–604
Sriraksa N, Wattanathorn J, Muchimapura S, Tiamkao S, Brown K, Chaisiwamongkol K (2012) Cognitive-enhancing effect of quercetin in a rat model of Parkinson’s disease induced by 6-hydroxydopamine. Evid Based Complement Alternat Med 2012:823206
Yao Y, Han DD, Zhang T, Yang Z (2010) Quercetin improves cognitive deficits in rats with chronic cerebral ischemia and inhibits voltage-dependent sodium channels in hippocampal CA1 pyramidal neurons. Phytother Res 24:136–140
Kumar A, Sehgal N, Kumar P, Padi SS, Naidu PS (2008) Protective effect of quercetin against ICV colchicine-induced cognitive dysfunctions and oxidative damage in rats. Phytother Res 22:1563–1569
Richetti SK, Blank M, Capiotti KM, Piato AL, Bogo MR, Vianna MR, Bonan CD (2011) Quercetin and rutin prevent scopolamine-induced memory impairment in zebrafish. Behav Brain Res 217:10–15
Sharma DR, Wani WY, Sunkaria A, Kandimalla RJ, Verma D, Cameotra SS, Gill KD (2013) Quercetin protects against chronic aluminium-induced oxidative stress and ensuing biochemical, cholinergic, and neurobehavioral impairments in rats. Neurotox Res 23:336–357
Vepsalainen S, Koivisto H, Pekkarinen E, Makinen P, Dobson G, McDougall GJ, Stewart D, Haapasalo A, Karjalainen RO, Tanila H, Hiltunen M (2013) Anthocyanin-enriched bilberry and blackcurrant extracts modulate amyloid precursor protein processing and alleviate behaviorial abnormalities in the APP/PS1 mouse model of Alzheimer’s disease. J Nutr Biochem 24:360–370
Ansari MA, Abdul HM, Joshi G, Opii WO, Butterfield DA (2009) Protective effect of quercetin in primary neurons against Abeta(1–42): relevance to Alzheimer’s disease. J Nutr Biochem 20:269–275
Garcia-Alloza M, Robbins EM, Zhang-Nunes SX, Purcell SM, Betensky RA, Raju S, Prada C, Greenberg SM, Bacskai BJ, Frosch MP (2006) Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol Dis 24:516–524
Zhang W, Hao J, Liu R, Zhang Z, Lei G, Su C, Miao J, Li Z (2011) Soluble Abeta levels correlate with cognitive deficits in the 12-month-old APPswe/PS1dE9 mouse model of Alzheimer’s disease. Behav Brain Res 222:342–350
Savonenko A, Xu GM, Melnikova T, Morton JL, Gonzales V, Wong MP, Price DL, Tang F, Markowska AL, Borchelt DR (2005) Episodic-like memory deficits in the APPswe/PS1dE9 mouse model of Alzheimer’s disease: relationships to beta-amyloid deposition and neurotransmitter abnormalities. Neurobiol Dis 18:602–617
Zong Y, Wang H, Dong W, Quan X, Zhu H, Xu Y, Huang L, Ma C, Qin C (2011) miR-29c regulates BACE1 protein expression. Brain Res 1395:108–115
Wang D, Li X, Gao K, Lu D, Zhang X, Ma C, Ye F, Zhang L (2013) Cardiotrophin-1 (CTF1) ameliorates glucose-uptake defects and improves memory and learning deficits in a transgenic mouse model of Alzheimer’s disease. Pharmacol Biochem Behav 107:48–57
Rinwa P, Kumar A (2013) Quercetin along with piperine prevents cognitive dysfunction, oxidative stress and neuro-inflammation associated with mouse model of chronic unpredictable stress. Arch Pharm Res. doi:10.1007/s12272-013-0205-4
Liu CM, Zheng GH, Cheng C, Sun JM (2013) Quercetin protects mouse brain against lead-induced neurotoxicity. J Agric Food Chem 61:7630–7635
Bevins RA, Besheer J (2006) Object recognition in rats and mice: a one-trial non-matching-to-sample learning task to study ‘recognition memory’. Nat Protoc 1:1306–1311
Takamura A, Okamoto Y, Kawarabayashi T, Yokoseki T, Shibata M, Mouri A, Nabeshima T, Sun H, Abe K, Urisu T, Yamamoto N, Shoji M, Yanagisawa K, Michikawa M, Matsubara E (2011) Extracellular and interneuronal HMW-AbetaOs represent a molecular basis of memory loss in Alzheimer’s disease model mouse. Mol Neurodegener 6:20
Laczo J, Vlcek K, Vyhnalek M, Vajnerova O, Ort M, Holmerova I, Tolar M, Andel R, Bojar M, Hort J (2009) Spatial navigation testing discriminates two types of amnestic mild cognitive impairment. Behav Brain Res 202:252–259
Liang KC, Hon W, Tyan YM, Liao WL (1994) Involvement of hippocampal NMDA and AMPA receptors in acquisition, formation and retrieval of spatial memory in the Morris water maze. Chin J Physiol 37:201–212
Kung MP, Hou C, Zhuang ZP, Zhang B, Skovronsky D, Trojanowski JQ, Lee VM, Kung HF (2002) IMPY: an improved thioflavin-T derivative for in vivo labelling of beta-amyloid plaques. Brain Res 956:202–210
Urbanc B, Cruz L, Le R, Sanders J, Ashe KH, Duff K, Stanley HE, Irizarry MC, Hyman BT (2002) Neurotoxic effects of thioflavin S-positive amyloid deposits in transgenic mice and Alzheimer’s disease. Proc Natl Acad Sci USA 99:13990–13995
Bussiere T, Bard F, Barbour R, Grajeda H, Guido T, Khan K, Schenk D, Games D, Seubert P, Buttini M (2004) Morphological characterization of Thioflavin-S-positive amyloid plaques in transgenic Alzheimer mice and effect of passive Abeta immunotherapy on their clearance. Am J Pathol 165:987–995
Dragicevic N, Mamcarz M, Zhu Y, Buzzeo R, Tan J, Arendash GW, Bradshaw PC (2010) Mitochondrial amyloid-beta levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer’s transgenic mice. J Alzheimers Dis 20(Suppl 2):S535–S550
Brown MR, Geddes JW, Sullivan PG (2004) Brain region-specific, age-related, alterations in mitochondrial responses to elevated calcium. J Bioenerg Biomembr 36:401–406
Schmidt R, Kienbacher E, Benke T, Dal-Bianco P, Delazer M, Ladurner G, Jellinger K, Marksteiner J, Ransmayr G, Schmidt H, Stogmann E, Friedrich J, Wehringer C (2008) Sex differences in Alzheimer’s disease. Neuropsychiatr 22:1–15
Tedeschi A, D’Errico G, Lauro MR, Sansone F, Di Marino S, D’Ursi AM, Aquino RP (2010) Effect of flavonoids on the Abeta(25–35)-phospholipid bilayers interaction. Eur J Med Chem 45:3998–4003
Wallace DC (2005) A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 39:359–407
Droge W (2002) Free radicals in the physiological control of cell function. Physiol Rev 82:47–95
Hagen TM, Yowe DL, Bartholomew JC, Wehr CM, Do KL, Park JY, Ames BN (1997) Mitochondrial decay in hepatocytes from old rats: membrane potential declines, heterogeneity and oxidants increase. Proc Natl Acad Sci USA 94:3064–3069
Trifunovic A, Larsson NG (2008) Mitochondrial dysfunction as a cause of ageing. J Intern Med 263:167–178
Sanz N, Diez-Fernandez C, Alvarez A, Cascales M (1997) Age-dependent modifications in rat hepatocyte antioxidant defence systems. J Hepatol 27:525–534
Sastre J, Pallardo FV, Pla R, Pellin A, Juan G, O’Connor JE, Estrela JM, Miquel J, Vina J (1996) Aging of the liver: age-associated mitochondrial damage in intact hepatocytes. Hepatology 24:1199–1205
Tillement L, Lecanu L, Yao W, Greeson J, Papadopoulos V (2006) The spirostenol (22R, 25R)-20alpha-spirost-5-en-3beta-yl hexanoate blocks mitochondrial uptake of Abeta in neuronal cells and prevents Abeta-induced impairment of mitochondrial function. Steroids 71:725–735
Schmidt C, Lepsverdize E, Chi SL, Das AM, Pizzo SV, Dityatev A, Schachner M (2008) Amyloid precursor protein and amyloid beta-peptide bind to ATP synthase and regulate its activity at the surface of neural cells. Mol Psychiatry 13:953–969
Hauptmann S, Scherping I, Drose S, Brandt U, Schulz KL, Jendrach M, Leuner K, Eckert A, Muller WE (2009) Mitochondrial dysfunction: an early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol Aging 30:1574–1586
Pagani L, Eckert A (2011) Amyloid-beta interaction with mitochondria. Int J Alzheimers Dis 2011:925050
Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, Casadesus G, Zhu X (2008) Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci USA 105:19318–19323
Schmitt K, Grimm A, Kazmierczak A, Strosznajder JB, Gotz J, Eckert A (2012) Insights into mitochondrial dysfunction: aging, amyloid-beta, and tau-A deleterious trio. Antioxid Redox Signal 16:1456–1466
Spuch C, Ortolano S, Navarro C (2012) New insights in the amyloid-beta interaction with mitochondria. J Aging Res 2012:324968
Fernandez-Vizarra P, Fernandez AP, Castro-Blanco S, Serrano J, Bentura ML, Martinez-Murillo R, Martinez A, Rodrigo J (2004) Intra- and extracellular Abeta and PHF in clinically evaluated cases of Alzheimer’s disease. Histol Histopathol 19:823–844
Manczak M, Mao P, Calkins MJ, Cornea A, Reddy AP, Murphy MP, Szeto HH, Park B, Reddy PH (2010) Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer’s disease neurons. J Alzheimers Dis 20(Suppl 2):S609–S631
Hardie DG (2011) AMP-activated protein kinase: a cellular energy sensor with a key role in metabolic disorders and in cancer. Biochem Soc Trans 39:1–13
Alba G, El Bekay R, Alvarez-Maqueda M, Chacon P, Vega A, Monteseirin J, Santa Maria C, Pintado E, Bedoya FJ, Bartrons R, Sobrino F (2004) Stimulators of AMP-activated protein kinase inhibit the respiratory burst in human neutrophils. FEBS Lett 573:219–225
Kukidome D, Nishikawa T, Sonoda K, Imoto K, Fujisawa K, Yano M, Motoshima H, Taguchi T, Matsumura T, Araki E (2006) Activation of AMP-activated protein kinase reduces hyperglycemias-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes 55:120–127
Xie Z, Zhang J, Wu J, Viollet B, Zou MH (2008) Upregulation of mitochondrial uncoupling protein-2 by the AMP-activated protein kinase in endothelial cells attenuates oxidative stress in diabetes. Diabetes 57:3222–3230
Calegari VC, Zoppi CC, Rezende LF, Silveira LR, Carneiro EM, Boschero AC (2011) Endurance training activates AMP-activated protein kinase, increases expression of uncoupling protein 2 and reduces insulin secretion from rat pancreatic islets. J Endocrinol 208:257–264
Yang YM, Han CY, Kim YJ, Kim SG (2010) AMPK-associated signalling to bridge the gap between fuel metabolism and hepatocyte viability. World J Gastroenterol 16:3731–3742
Shin SM, Kim SG (2009) Inhibition of arachidonic acid and iron-induced mitochondrial dysfunction and apoptosis by oltipraz and novel 1,2-dithiole-3-thione congeners. Mol Pharmacol 75:242–253
Dong GZ, Jang EJ, Kang SH, Cho IJ, Park SD, Kim SC, Kim YW (2013) Red ginseng abrogates oxidative stress via mitochondria protection mediated by LKB1-AMPK pathway. BMC Complement Altern Med 13:64
Choi SH, Kim YW, Kim SG (2010) AMPK-mediated GSK3beta inhibition by isoliquiritigenin contributes to protecting mitochondria against iron-catalyzed oxidative stress. Biochem Pharmacol 79:1352–1362
Shin SM, Cho IJ, Kim SG (2009) Resveratrol protects mitochondria against oxidative stress through AMP-activated protein kinase-mediated glycogen synthase kinase-3beta inhibition downstream of poly(ADP-ribose)polymerase-LKB1 pathway. Mol Pharmacol 76:884–895
Ng CH, Guan MS, Koh C, Ouyang X, Yu F, Tan EK, O’Neill SP, Zhang X, Chung J, Lim KL (2012) AMP kinase activation mitigates dopaminergic dysfunction and mitochondrial abnormalities in Drosophila models of Parkinson’s disease. J Neurosci 32:14311–14317
Moran C, Sanz-Rodriguez A, Jimenez-Pacheco A, Martinez-Villareal J, McKiernan RC, Jimenez-Mateos EM, Mooney C, Woods I, Prehn JH, Henshall DC, Engel T (2013) Bmf upregulation through the AMP-activated protein kinase pathway may protect the brain from seizure-induced cell death. Cell Death Dis 4:e606
Wang L, Brautigan DL (2013) Alpha-SNAP inhibits AMPK signalling to reduce mitochondrial biogenesis and dephosphorylates Thr172 in AMPK alpha in vitro. Nat Commun 4:1559
Finocchietto PV, Holod S, Barreyro F, Peralta JG, Alippe Y, Giovambattista A, Carreras MC, Poderoso JJ (2011) Defective leptin-AMP-dependent kinase pathway induces nitric oxide release and contributes to mitochondrial dysfunction and obesity in ob/ob mice. Antioxid Redox Signal 15:2395–2406
Reddy PH, Tripathi R, Troung Q, Tirumala K, Reddy TP, Anekonda V, Shirendeb UP, Calkins MJ, Reddy AP, Mao P, Manczak M (2012) Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer’s disease: implications to mitochondria-targeted antioxidant therapeutics. Biochim Biophys Acta 1822:639–649
Eckert GP, Renner K, Eckert SH, Eckmann J, Hagl S, Abdel-Kader RM, Kurz C, Leuner K, Muller WE (2012) Mitochondrial dysfunction: a pharmacological target in Alzheimer’s disease. Mol Neurobiol 46:136–150
Won JS, Im YB, Kim J, Singh AK, Singh I (2010) Involvement of AMP-activated-protein-kinase (AMPK) in neuronal amyloidogenesis. Biochem Biophys Res Commun 399:487–491
Vingtdeux V, Giliberto L, Zhao H, Chandakkar P, Wu Q, Simon JE, Janle EM, Lobo J, Ferruzzi MG, Davies P, Marambaud P (2010) AMP-activated protein kinase signalling activation by resveratrol modulates amyloid-beta peptide metabolism. J Biol Chem 285:9100–9113
Lu J, Wu DM, Zheng YL, Hu B, Zhang ZF, Shan Q, Zheng ZH, Liu CM, Wang YJ (2010) Quercetin activates AMP-activated protein kinase by reducing PP2C expression protecting old mouse brain against high cholesterol-induced neurotoxicity. J Pathol 222:199–212
Greco SJ, Hamzelou A, Johnston JM, Smith MA, Ashford JW, Tezapsidis N (2011) Leptin boosts cellular metabolism by activating AMPK and the sirtuins to reduce tau phosphorylation and beta-amyloid in neurons. Biochem Biophys Res Commun 414:170–174
Greco SJ, Sarkar S, Johnston JM, Tezapsidis N (2009) Leptin regulates tau phosphorylation and amyloid through AMPK in neuronal cells. Biochem Biophys Res Commun 380:98–104
Vingtdeux V, Chandakkar P, Zhao H, d’Abramo C, Davies P, Marambaud P (2011) Novel synthetic small-molecule activators of AMPK as enhancers of autophagy and amyloid-beta peptide degradation. FASEB J 25:219–231
Kwon KJ, Kim HJ, Shin CY, Han SH (2010) Melatonin Potentiates the Neuroprotective Properties of Resveratrol Against Beta-Amyloid-Induced Neurodegeneration by Modulating AMP-Activated Protein Kinase Pathways. J Clin Neurol 6:127–137
Acknowledgments
The present work was supported by National Natural Science Foundation of China (U1304806) and the Scientific Research Fund of Henan University of Science and Technology (NO. 09001664).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Wang, DM., Li, SQ., Wu, WL. et al. Effects of Long-Term Treatment with Quercetin on Cognition and Mitochondrial Function in a Mouse Model of Alzheimer’s Disease. Neurochem Res 39, 1533–1543 (2014). https://doi.org/10.1007/s11064-014-1343-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-014-1343-x