
Abstract
Phosphorylated tau was found to be regulated after cerebral ischemia and linked to high risk for the development of post-stroke dementia. Our previous study showed that ginsenoside Rd (Rd), one of the main active ingredients in Panax ginseng, decreased tau phosphorylation in Alzheimer model. As an extending study, here we investigated whether Rd could reduce tau phosphorylation and sequential cognition impairment after ischemic stroke. Sprague–Dawley rats were subjected to focal cerebral ischemia. The tau phosphorylation of rat brains were analyzed following ischemia by Western blot and animal cognitive functions were examined by Morris water maze and Novel object recognition task. Ischemic insults increased the levels of phosphorylated tau protein at Ser199/202 and PHF-1 sites and caused animal memory deficits. Rd treatment attenuated ischemia-induced enhancement of tau phosphorylation and ameliorated behavior impairment. Furthermore, we revealed that Rd inhibited the activity of Glycogen synthase kinase-3β (GSK-3β), the most important kinase involving tau phosphorylation, but enhanced the activity of protein kinase B (PKB/AKT), a key kinase suppressing GSK-3β activity. Moreover, we found that LY294002, an antagonist for phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathway, abolished the inhibitory effect of Rd on GSK-3β activity and tau phosphorylation. Taken together, our findings provide the first evidence that Rd may reduce cerebral ischemia-induced tau phosphorylation via the PI3K/AKT/GSK-3β pathway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ischemic stroke is a major cause of mortality and disability in human and the second leading cause of death in developed countries [1]. Apart from physical disability, post-stroke dementia (PSD) is the second most common consequence caused by stroke [2]. Epidemiological studies have shown that the incidence of PSD is about 32 % 1 year after stroke and 12 times higher than in normal population 4 years after stroke [3, 4]. PSD is associated with a high mortality rate in stroke patients and seriously affects the quality of their lives [3, 4]. Growing evidence showed that many of these dementias developed progressively and the primary damage was not the direct cause of the PSD in most of the cases [5].
It is reported that PSD shares some common risk factors and neuropathology features with a well-known Alzheimer’s disease (AD), e.g. formation of neurofibrillary tangles (NFTs) mainly composed of hyperphosphorylated tau protein [6–9]. Tau protein is microtubule-associated proteins that is expressed abundantly in central nervous system (CNS) [10]. It induces microtubule assembly and stabilizes microtubules by its phosphorylation states which are mainly regulated by phosphatidylinositol 3-kinase/protein kinase B/Glycogen synthase kinase-3β (PI3K/AKT/GSK-3β) pathway [11–16]. However, hyperphosphorylation of tau forms NFTs and leads to dysfunction of synapses, degeneration of neurons, and cognitive impairment [10, 17]. Therefore, inhibition of tau phosphorylation may be a potential therapeutic strategy for preventing such neurodegenerative diseases [18].
Ginsenosides are the most active ingredients in ginseng which has been widely used as a traditional herbal medicine for thousands of years in eastern Asia [19]. Our pre-clinical and clinical studies showed that ginsenoside Rd (Rd), a monomer compound extracted from ginseng, was effective and safe for the treatment of acute ischemic stroke [20–27], suggesting that Rd may be a promising neuroprotectant. Moreover, our latest studies showed that Rd inhibited tau phosphorylation and ameliorated cognitive function in rat model of AD [28–30]. Thus, as an extending study, here we investigated whether Rd could affect experimental cerebral ischemia-induced tau phosphorylation and sequential cognition dysfunction and further explored the underlying mechanisms in this process.
Materials and Methods
Materials
Rd with a purity of 98 % was obtained from Tai–He Biopharmaceutical Co. Ltd. (Guangzhou, China). The stock solutions were prepared in saline containing 10 % 1,3-propanediol (v/v). cresyl violet and 2,3,5-triphenyltetrazolium chloride (TTC) were purchased from Sigma-Aldrich Inc. (St. Louis, Mo, USA). PHF-1 antibody was obtained from Abcam (Cambridge, UK) and other antibodies were purchased from Cell Signaling (Carlsbad, CA, USA). All other reagents were from commercial suppliers and of standard biochemical quality.
Focal Cerebral Ischemia
Male Sprague–Dawley rats weighing 270–320 g, provided by the Animal Breeding Center, affiliated to the Fourth Military Medical University, China, were used in this study. Animal protocols were approved by the Ethics Committee for Animal Experimentation of the Fourth Military Medical University. The focal cerebral ischemia was induced by 2 h of middle cerebral artery occlusion (MCAO) as described previously with modifications [24]. In brief, animals were anesthetized with a mixture of isoflurane (1.5–2 %), oxygen and nitrogen. Body temperature in the rectum was maintained at 37.0 °C using a thermostatically controlled heating blanket. A 4–0 monofilament coated with poly-l-lysine was introduced through the internal carotid artery to occlude the origin of the middle cerebral artery (MCA). The induction of focal cerebral ischemia was verified with laser Doppler flowmetry (PeriFlux 5000; Perimed AB, Sweden). A drop in regional cerebral blood flow (CBF) below 30 % from baseline after the insertion of the filament was considered to be sufficient for induction of focal cerebral ischemia. Control animals were subjected to the same surgical procedures except that the suture was not advanced into the MCA. Rd with a concentration of 30 mg/kg or vehicle was applied intraperitoneally 1 h before MCAO and 10 mg/kg per day until animals were sacrificed.
Nissl Staining
Nissl staining was performed as we previously described with some modifications [30]. Briefly, the sections of brains were placed in xylene twice (15 min/every time), then placed in graded alcohols (100, 90, 80, 70, 50 %, 5 min/every time), immersed in 0.1 % cresyl violet 5 min, dehydrated through graded alcohols (95, 100 %, 2 min/every time), placed in xylene twice again (15 min/every time) and mounted by neutral gum, the numbers of remaining neurons in ipsilateral Hippocampal CA1 region were calculated for each experimental group.
TTC Staining
TTC stain was performed as we previously described [26]. Briefly, rats in each group were anesthetized and decapitated. Each brain was sliced into 2 mm sections. Serial coronal sections were soaked in 2 % TTC phosphate buffer at 37 °C for 10 min in the dark. Normal brain tissues were stained red, while infarct tissues were not stained (white). The sections were soaked in 4 % paraformaldehyde phosphate buffer for 30 min, arranged in order and scanned. The ratios percentages of infarct areas to the total brain areas were calculated.
Novel Object Recognition (NOR)
The NOR takes advantage of the natural tendency of rats to explore a novel object more than a familiar object and was used to evaluate the working memory from post-occlusion day (POD) 26 to POD 32 as previously described [31]. Briefly, rats were trained in the experimental chamber with two identical objects. They were allowed to explore the objects for 10 min. Memory retention was assessed either 1 h (for short-term retention) or 24 h (for long-term retention) after training. The animals were allowed to explore two different objects for 5 min. One of the objects was identical to those explored during the training session (familiar object) and the other was a different object (novel object). The animals were regarded to be exploring when they were facing, sniffing, or biting the object. The time spent exploring object was recorded and analyzed using ANY-maze software (Stoeling Co, Wood Dale, IL, USA). The relative time of novel object exploration was calculated as the index of discrimination calculated as follows: (Tnovel − Tfamiliar)/(Tnovel + Tfamiliar). The total times of exploration for each group in training and testing sessions were compared to verify that there were no differences between groups in this parameter. Animals showing low exploration times were excluded from the experiments.
Morris Water Maze
To assess the spatial memory deficit of rats after MCAO, Morris water maze (MWM), including 5 days of spatial acquisition and 1 day of probe trial (POD 26–32), was performed as we previously described [32]. Rats were individually trained in a circular pool (150 cm diameter, 60 cm height) filled with water (22 °C). The platform was submerged about 1.5 cm below the surface of the water infiltrated with 80 ml Chinese ink. On each day, the animal was subjected to three trials and each trial lasted either until the rat found the platform or for 120 s. Time to reach the platform (latency), length of swim path, and swim speed were recorded semi-automatically by a video tracking system. 24 h after the last learning session, the platform was removed from its previous location and the animals were given a probe trial in which they had 60 s to search for the platform. Time spent in the target quadrant, number of times animals crossed above the former target site where the platform had been located (crossovers) were recorded during the probe trials. To control for possible differences in visual acuity or sensorimotor function between groups, a cued task were performed using a visible platform raised 0.5 cm above the surface of the water.
Primary Culture of Neurons
Neurons were cultured from Sprague–Dawley rat embryos as previously described with some modifications [23]. Animal procedures were in accordance with NIH Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80-23, revised 1996). All efforts were made to minimize animal number and their suffering. After trituration and trypsinization, the single-cell suspension from embryos Hippocampus was seeded in 6-well plates coated with poly-l-lysine (50 μg/ml) at the density of (2–2.5) × 105/cm2. Cells were maintained in Neurobasal medium supplemented with 2 % B-27, 0.5 mM l-glutamine, 100 U/ml penicillin and 100 mg/ml streptomycin in a humidified atmosphere of 5 % CO2 at 37 °C.
Oxygen–Glucose Deprivation (OGD)
OGD was carried out as described by Li et al. [22]. Briefly, the culture medium was replaced with pre-warmed Dulbecco’s modified eagle medium (DMEM) without glucose and serum. The cell cultures were then transferred into an anaerobic chamber equilibrated with 95 % N2 and 5 % CO2. The chamber was kept in a 37 °C incubator. Control cultures were maintained in a normal oxygenated DMEM containing 25 mM glucose. After 2 h, cultures were placed back to the normoxic incubator with normal culture medium. LY294002 (5 μM) was pretreated 12 h before OGD. Rd (10 μM) was added in the culture medium during OGD and reoxygenation.
Western Blot
Ipsilateral hippocampus after MCAO was homogenized on ice in the RIPA lysis buffer (Beyotime, China) containing 0.5 mM PMSF. Western blot was performed according to Ye R et al. [24]. Briefly, protein samples were electrophoresed on a 10 % SDS-PAGE and subsequently transferred to PVDF membrane (Millipore, USA). The membrane was incubated in blocking buffer containing 5 % non-fat dried milk at room temperature for 1 h and then probed with the primary antibody (PHF-1 at 1:500; p-AKT at 1:500; tau-5 at 1:1,500; AKT at 1:2,000; S199/202 at 1:500 GSK-3β at 1:1,000; p-GSK-3β at 1:500 and GAPDH at 1:3,000) in blocking buffer at 4 °C overnight. The membrane was washed three times with TBST (TBS and 0.1 % Tween 20) and then incubated with HRP-conjugated secondary antibody at room temperature for another 1 h. Specific signals of proteins were visualized by chemiluminescence using the ECL western blotting detection system (GE Healthcare UK). For quantitative analysis, the ratio of the specific signals of protein (relative intensity of the signal) to that of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) protein was calculated.
Statistical Analysis
Results were analyzed with one-way ANOVA followed by Tukey post test for multiple comparisons. Origin-pro 8 software was used for statistical tests. Statistical significance was established at p < 0.05.
Results
Rd Reduces Infarction Volume and Hippocampal Cell Loss After MCAO
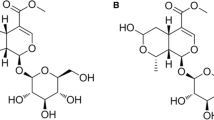
Ginsenoside Rd (Dammar-24[25]-ene-3,12,20[S]-triol-]20-O-β-d-gluco-pyranosyl]-3-O-β-d-glucopyranosyl-(1 → 2)-β-dglucopyranoside[Rd]; Fig. 1a) is one of the major ginsenosides in the ginseng root [27]. Rd has exhibited an encouraging neuroprotective efficacy against cerebral ischemic stroke in both laboratory and clinical studies [20–27]. Consistently, in this study we showed that Rd administration could reduce infarction volume POD 1 after MCAO, as revealed by TTC and nissl staining (Fig. 1b, c), we also found that Rd could decrease cell loss in ipsilateral hippocampal CA1 region POD 28 after MCAO (Fig. 1d), these results suggested that Rd was effective in the treatment of ischemic stroke.

The neuroprotective effect of Rd after rat MCAO. Infarction sizes were evaluated by TTC (b) and Nissl staining (c) 1 day after MCAO. a The structural formulae of Rd. b TTC staining of infarcts in brain sections (white areas marked by number sign). c Nissl staining of infarcts in brain sections (boxed by black lines). (d) Nissl staining of cell loss in ipsilateral hippocampal CA1 region 28 days after MCAO. Data are expressed as mean ± SEM (n = 9). Mean values in Sham groups were scaled to 100 % (Nissl staining). * p < 0.05 versus MCAO + Rd group
Rd Improves Cognitive Function After MCAO
NOR and MWM were respectively performed to assess the effects of Rd on working and spatial memory after ischemic brain injury. For working memory, the rats subjected to MCAO injury spent less time exploring the novel objects, compared with the sham group. Relative to the MCAO group, the rats in Rd-treated group spent a greater proportion of time exploring the novel objects in either 1 h or 24 h tests (Fig. 2a, b). For spatial learning and memory, mean latency of finding the platform and the path length to reach the platform declined progressively during the training period in sham rats. By contrast, the rats in MCAO group had more time of latency and longer distance of path length. Less time in target quadrant and platform crossovers was also found in MCAO group. However, these changes were markedly attenuated by Rd administration (Fig. 2c–f). In addition, all the rats could swim easily in all three groups and swimming speed of three groups did not differ during pre-training (data not shown). These results above indicated that Rd could improve working memory and spatial learning function after ischemic brain injury.

Effects of Rd on cognitive function after rat MCAO. Learning and memory performance were evaluated in novel object recognition (NOR) (a, b) and Morris water maze (MWM) (c–f) task 28 days after MCAO. Performance on object recognition was tested 1 h (a) and 24 h (b) after training with two identical objects. In MWM task, spatial acquisition trial [path length (c), escape latency (d)] and probe trial [time in garget quadrant (e), platform crossovers (f)] were performed in order to detect spatial learning and memory ability. Data are expressed as mean ± SEM (n = 14). * p < 0.05, ** p < 0.01 versus MCAO group
Rd Attenuates Tau Protein Phosphorylation After MCAO
Hyperphosphorylated tau is highly associated with cognitive impairment and has been found in the ischemic brain [6–11, 17, 33, 34]. To determine the effects of Rd on the level of phosphorylated tau (p-tau) in vivo, we investigated the expression of total tau (tau-5) and p-tau (PHF-1 and S199/202) 24 h/7 days/14 days and 28 days after MCAO (Fig. 3, Fig. S1). Western blotting results showed that PHF-1 and S199/202 levels increased in all of these time points, but tau-5 was not affected after MCAO, consistent with previous reports [33, 34]. Rd treatment significantly reduced MCAO-induced increase of p-tau levels at these time points (Fig. 3a–d, Fig. S1a-d) but did not affect tau-5 expression (Fig. 3e, f, Fig. S1e-f). These results indicated that Rd could attenuate tau protein phosphorylation after ischemic injury.

Effects of Rd on p-tau expression in ischemic rat brain 24 h and 28 days after MCAO. Western blotting analysis was performed using antibodies against S199/202 (a 24 h and b 28 days) or PHF-1 (c 24 h and d 28 days) or tau-5 (e 24 h and f 28 days) as described in Methods. Data are expressed as mean ± SEM (n = 6). GAPDH was used as internal control. Mean values in vehicle-treated (Sham) groups were scaled to 100 %. * p < 0.05 versus MCAO group, SA saline
Rd Enhances GSK-3β and AKT Phosphorylation After MCAO
GSK-3β is one of the most important kinases involving in tau phosphorylation in the brain [12–16] and can be inhibited by AKT, a kinase involving in survival signaling pathways [35–37]. We then explored whether Rd could affect PI3K/AKT/GSK-3β pathway in MCAO rats (Fig. 4). The activities of GSK-3β and AKT kinases are dependent on their phosphorylation states. To be phosphorylated inhibits GSK-3β (at Ser9 site) but activates AKT. Western blotting results showed that the levels of p-GSK-3β and p-AKT were reduced 24 h after rat MCAO (Fig. 4a, c) and returned to normal levels 28 days after surgery (Fig. 4b, d), MCAO also changed p-GSK-3β and p-AKT levels at POD 7 and POD 14 (Fig. S2), these results were consistent with previous reports [38]. With the treatment of Rd, the levels of p-AKT and p-GSK-3β were markedly increased in these time points, indicating that Rd may suppress GSK-3β activity but enhance AKT activity.

Effects of Rd on AKT and GSK-3β phosphorylation in ischemic rat brain after MCAO. Western blotting analysis was performed using antibodies against p-GSK-3β/GSK-3β (a 24 h and b 28 days) or p-AKT/AKT (c 24 h and d 28 days) as described in Methods. Data are expressed as mean ± SEM (n = 6). Mean values in vehicle-treated (Sham) groups were scaled to 100 %. * p < 0.05 versus MCAO + Rd group, SA saline
Rd Affects Tau Phosphorylation Via PI3K/AKT/GSK-3β Pathway
Since AKT and GSK-3β are key participants in the PI3K/AKT/GSK-3β signaling pathway, we then investigated whether Rd affected tau phosphorylation via this pathway by using a PI3K/AKT pathway inhibitor LY294002 on MCAO rats and primary cultured neurons exposed to OGD injury. Western blotting results showed that compared with the sham group, MCAO increased p-tau but decreased p-GSK-3β levels, which was attenuated by Rd administration (Fig. 5a, b, d), consistent with our previous results (Fig. 4). With the treatment of LY294002, the inhibitory effects of Rd on tau phosphorylation and GSK-3β activity were significantly blocked (Fig. 5a, b, d). Our in vitro study also drew a similar conclusion. That is, Rd could reduce OGD induced tau phosphorylation and GSK-3β activation, but these effects were abolished by LY294002 administration (Fig. S3). It was noted that tau-5 expression was not affected with the treatment of Rd or LY294002 (Fig. 5c).

PI3K/AKT/GSK-3β pathway is involved in the effect of Rd on tau phosphorylation in vivo. Western blotting analysis of proteins extracted from ischemic rat brain 1 day after MCAO was performed using antibodies against S199/202 (a) or PHF-1 (b) or tau-5 (c) or p-GSK-3β (d) as described in Methods. LY294002 (LY, 5 g/l, 5 μl, i.c.v.) was pretreated 1 h before MCAO. Data are expressed as mean ± SEM (n = 6). GAPDH was used as internal control. Mean values in sham-treated groups were scaled to 100 %. * p < 0.05 versus MCAO + Rd group, SA saline
Discussion
In the present study, we investigated the effects of Rd on tau protein phosphorylation as well as cognitive function in MCAO rats and showed that Rd attenuated cognitive impairment and decreased the levels of p-tau protein after ischemic injury. Moreover, Rd could enhance AKT and GSK-3β phosphorylation, which was blocked by PI3K/AKT inhibitor LY294002, suggesting that PI3K/AKT/GSK-3β signaling pathway was involved in the inhibitory effects of Rd on tau protein phosphorylation, which may account for cognitive function improvement after Rd administration.
Ischemic stroke not only cause physical disability, but also PSD [2]. In community-based studies, the prevalence of dementia in people with a history of stroke was about 5.8-times higher than in those who have not had stroke [39, 40]. In hospital-based studies, the incidence of PSD ranges from 5.9 to 32 % [3]. Ischemic stroke-induced progressive cognitive impairment was also confirmed in animal model of stroke [41–43]. The mechanism underlying PSD is still unclear. Tatemichi et al. [44] reported that more than 60 % of PSD patients developed AD pathology. Many pre-clinical studies also found that focal cerebral ischemia induced AD-like pathological changes in rodent model [8, 33, 34, 45]. Consistently, the present study showed MCAO injury caused cognitive deficits and tau protein phosphorylation (Figs. 2, 3).
Preventing tau hyperphosphorylation was proposed to be a potential therapeutic strategy for AD-like diseases [18]. Our previous study showed that Rd could inhibited tau phosphorylation in rat model of AD [28–30]. As an extending study, the present study showed that daily administration of Rd could also inhibit tau phosphorylation and ameliorate cognitive function after experimental cerebral ischemia in rats (Figs. 2, 3).
The PI3K/AKT/GSK-3β pathway is pivotal to maintenance of the neuronal network, cell survival, and longevity. Dysregulation of this signaling pathway is the major mechanism underlying the pathology of neurodegenerative diseases, such as AD [46]. GSK-3β is a major tau kinase playing a crucial role in AD-like tau hyperphosphorylation [12–16] and its activity can be inhibited by AKT at Ser9-phosphorylation site [46]. Our present study showed that AKT was inactivated but GSK-3β was activated after MCAO (Fig. 4), consistent with a previous study [38], and these results may partly explain why p-tau levels increased after stroke. Furthermore we showed that Rd inhibited GSK-3β but enhanced AKT activities whereas PI3K/AKT inhibitor LY294002 could block this effect of Rd (Fig. 5), suggesting that Rd may affect tau phosphorylation via PI3K/AKT/GSK-3β pathway.
Estrogen receptor (ER)-mediated PI3K/AKT activation is well-documented in protection against cell death. Ginsenosides were structurally and functionally similar to 17β-estradiol and activated AKT pathways [47, 48]. Thus, we proposed that acting on ER may be responsible for Rd-induced AKT activation and sequential p-Tau down-regulation. Yet, in our previous studies Rd was showed to prevent from glutamate/oxygen–glucose deprivation (OGD)-induced apoptosis in cultured neurons and reduce infarction volume after transient focal ischemia in rats [20–27], Rd could also increase Glutamate transporter-1 (GLT-1) expression in astrocytes for glutamate clearance [49]. Thus, apart from reducing p-Tau levels, other mechanisms may involve in the ameliorate effects of Rd on cognitive function after cerebral ischemic injury as well. Further investigation is still needed.
In conclusion, in the present study we provide the first evidence that Rd could improve cognitive function and reduce tau protein phosphorylation via the PI3K/AKT/GSK-3β pathway after experimental cerebral ischemia, indicating that Rd may serve as a promising drug for the treatment of PSD.
References
Goldstein LB, Adams R, Alberts MJ, Appel LJ, Brass LM, Bushnell CD, Culebras A, DeGraba TJ, Gorelick PB, Guyton JR, Hart RG, Howard G, Kelly-Hayes M, Nixon JV, Sacco RL (2006) Primary prevention of ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council: cosponsored by the Atherosclerotic Peripheral Vascular Disease Interdisciplinary Working Group; Cardiovascular Nursing Council; Clinical Cardiology Council; Nutrition, Physical Activity, and Metabolism Council; and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation 113(24):e873–e923. doi:10.1161/01.STR.0000223048.70103.F1
Murray CJ, Lopez AD (1997) Global mortality, disability, and the contribution of risk factors: Global Burden of Disease Study. Lancet 349(9063):1436–1442. doi:10.1016/S0140-6736(96)07495-8
Leys D, Henon H, Mackowiak-Cordoliani MA, Pasquier F (2005) Poststroke dementia. Lancet Neurol 4(11):752–759. doi:10.1016/S1474-4422(05)70221-0
Liu R, Yuan H, Yuan F, Yang SH (2012) Neuroprotection targeting ischemic penumbra and beyond for the treatment of ischemic stroke. Neurol Res 34(4):331–337. doi:10.1179/1743132812Y.0000000020
Tatemichi TK, Paik M, Bagiella E, Desmond DW, Stern Y, Sano M, Hauser WA, Mayeux R (1994) Risk of dementia after stroke in a hospitalized cohort: results of a longitudinal study. Neurology 44(10):1885–1891
de la Torre JC (2004) Is Alzheimer’s disease a neurodegenerative or a vascular disorder? Data, dogma, and dialectics. Lancet Neurol 3(3):184–190. doi:10.1016/S1474-4422(04)00683-0
White L, Petrovitch H, Hardman J, Nelson J, Davis DG, Ross GW, Masaki K, Launer L, Markesbery WR (2002) Cerebrovascular pathology and dementia in autopsied Honolulu–Asia Aging Study participants. Ann N Y Acad Sci 977:9–23
Zhang Q, Gao T, Luo Y, Chen X, Gao G, Gao X, Zhou Y, Dai J (2012) Transient focal cerebral ischemia/reperfusion induces early and chronic axonal changes in rats: its importance for the risk of Alzheimer’s disease. PLoS ONE 7(3):e33722. doi:10.1371/journal.pone.0033722
Iwata N, Higuchi M, Saido TC (2005) Metabolism of amyloid-beta peptide and Alzheimer’s disease. Pharmacol Ther 108(2):129–148. doi:10.1016/j.pharmthera.2005.03.010
Kanemaru K (2013) Immunotherapy targeting misfolded proteins in neurodegenerative disease. Brain Nerve 65(4):469–474
Cleveland DW, Hwo SY, Kirschner MW (1977) Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J Mol Biol 116(2):227–247
Billingsley ML, Kincaid RL (1997) Regulated phosphorylation and dephosphorylation of tau protein: effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem J 323(Pt 3):577–591
Noble W, Olm V, Takata K, Casey E, Mary O, Meyerson J, Gaynor K, LaFrancois J, Wang L, Kondo T, Davies P, Burns M, Veeranna NR, Dickson D, Matsuoka Y, Ahlijanian M, Lau LF, Duff K (2003) Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron 38(4):555–565
Morioka M, Kawano T, Yano S, Kai Y, Tsuiki H, Yoshinaga Y, Matsumoto J, Maeda T, Hamada J, Yamamoto H, Fukunaga K, Kuratsu J (2006) Hyperphosphorylation at serine 199/202 of tau factor in the gerbil hippocampus after transient forebrain ischemia. Biochem Biophys Res Commun 347(1):273–278. doi:10.1016/j.bbrc.2006.06.096
Reynolds CH, Betts JC, Blackstock WP, Nebreda AR, Anderton BH (2000) Phosphorylation sites on tau identified by nanoelectrospray mass spectrometry: differences in vitro between the mitogen-activated protein kinases ERK2, c-Jun N-terminal kinase and P38, and glycogen synthase kinase-3beta. J Neurochem 74(4):1587–1595
Engel T, Hernandez F, Avila J, Lucas JJ (2006) Full reversal of Alzheimer’s disease-like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase-3. J Neurosci 26(19):5083–5090. doi:10.1523/JNEUROSCI.0604-06.2006
Zhu LQ, Wang SH, Liu D, Yin YY, Tian Q, Wang XC, Wang Q, Chen JG, Wang JZ (2007) Activation of glycogen synthase kinase-3 inhibits long-term potentiation with synapse-associated impairments. J Neurosci 27(45):12211–12220. doi:10.1523/JNEUROSCI.3321-07.2007
Cancino GI, Toledo EM, Leal NR, Hernandez DE, Yevenes LF, Inestrosa NC, Alvarez AR (2008) STI571 prevents apoptosis, tau phosphorylation and behavioural impairments induced by Alzheimer’s beta-amyloid deposits. Brain 131(Pt 9):2425–2442. doi:10.1093/brain/awn125
Radad K, Gille G, Liu L, Rausch WD (2006) Use of ginseng in medicine with emphasis on neurodegenerative disorders. J Pharmacol Sci 100(3):175–186
Liu X, Xia J, Wang L, Song Y, Yang J, Yan Y, Ren H, Zhao G (2009) Efficacy and safety of ginsenoside-Rd for acute ischaemic stroke: a randomized, double-blind, placebo-controlled, phase II multicenter trial. Eur J Neurol 16(5):569–575. doi:10.1111/j.1468-1331.2009.02534.x
Liu X, Wang L, Wen A, Yang J, Yan Y, Song Y, Ren H, Wu Y, Li Z, Chen W, Xu Y, Li L, Xia J, Zhao G (2012) Ginsenoside-Rd improves outcome of acute ischaemic stroke—a randomized, double-blind, placebo-controlled, multicenter trial. Eur J Neurol 19(6):855–863. doi:10.1111/j.1468-1331.2011.03634.x
Li XY, Liang J, Tang YB, Zhou JG, Guan YY (2010) Ginsenoside Rd prevents glutamate-induced apoptosis in rat cortical neurons. Clin Exp Pharmacol Physiol 37(2):199–204. doi:10.1111/j.1440-1681.2009.05286.x
Ye R, Li N, Han J, Kong X, Cao R, Rao Z, Zhao G (2009) Neuroprotective effects of ginsenoside Rd against oxygen-glucose deprivation in cultured hippocampal neurons. Neurosci Res 64(3):306–310. doi:10.1016/j.neures.2009.03.016
Ye R, Yang Q, Kong X, Han J, Zhang X, Zhang Y, Li P, Liu J, Shi M, Xiong L, Zhao G (2011) Ginsenoside Rd attenuates early oxidative damage and sequential inflammatory response after transient focal ischemia in rats. Neurochem Int 58(3):391–398. doi:10.1016/j.neuint.2010.12.015
Ye R, Zhang X, Kong X, Han J, Yang Q, Zhang Y, Chen Y, Li P, Liu J, Shi M, Xiong L, Zhao G (2011) Ginsenoside Rd attenuates mitochondrial dysfunction and sequential apoptosis after transient focal ischemia. Neuroscience 178:169–180. doi:10.1016/j.neuroscience.2011.01.007
Ye R, Kong X, Yang Q, Zhang Y, Han J, Li P, Xiong L, Zhao G (2011) Ginsenoside rd in experimental stroke: superior neuroprotective efficacy with a wide therapeutic window. Neurotherapeutics 8(3):515–525. doi:10.1007/s13311-011-0051-3
Ye R, Zhao G, Liu X (2013) Ginsenoside Rd for acute ischemic stroke: translating from bench to bedside. Expert Rev Neurother 13(6):603–613. doi:10.1586/ern.13.51
Li L, Liu J, Yan X, Qin K, Shi M, Lin T, Zhu Y, Kang T, Zhao G (2011) Protective effects of ginsenoside Rd against okadaic acid-induced neurotoxicity in vivo and in vitro. J Ethnopharmacol 138(1):135–141. doi:10.1016/j.jep.2011.08.068
Li L, Liu Z, Liu J, Tai X, Hu X, Liu X, Wu Z, Zhang G, Shi M, Zhao G (2013) Ginsenoside Rd attenuates beta-amyloid-induced tau phosphorylation by altering the functional balance of glycogen synthase kinase 3beta and protein phosphatase 2A. Neurobiol Dis 54:320–328. doi:10.1016/j.nbd.2013.01.002
Liu J, Yan X, Li L, Zhu Y, Qin K, Zhou L, Sun D, Zhang X, Ye R, Zhao G (2012) Ginsennoside rd attenuates cognitive dysfunction in a rat model of Alzheimer’s disease. Neurochem Res 37(12):2738–2747. doi:10.1007/s11064-012-0866-2
Bevins RA, Besheer J (2006) Object recognition in rats and mice: a one-trial non-matching-to-sample learning task to study ‘recognition memory’. Nat Protoc 1(3):1306–1311. doi:10.1038/nprot.2006.205
Li N, Kong X, Ye R, Yang Q, Han J, Xiong L (2011) Age-related differences in experimental stroke: possible involvement of mitochondrial dysfunction and oxidative damage. Rejuvenation Res 14(3):261–273. doi:10.1089/rej.2010.1115
Zhang ZH, Xi GM, Li WC, Ling HY, Qu P, Fang XB (2010) Cyclic-AMP response element binding protein and tau are involved in the neuroprotective mechanisms of nerve growth factor during focal cerebral ischemia/reperfusion in rats. J Clin Neurosci 17(3):353–356. doi:10.1016/j.jocn.2009.07.086
Gordon-Krajcer W, Kozniewska E, Lazarewicz JW, Ksiezak-Reding H (2007) Differential changes in phosphorylation of tau at PHF-1 and 12E8 epitopes during brain ischemia and reperfusion in gerbils. Neurochem Res 32(4–5):729–737. doi:10.1007/s11064-006-9199-3
Kumar P, Miller AI, Polverini PJ (2004) p38 MAPK mediates gamma-irradiation-induced endothelial cell apoptosis, and vascular endothelial growth factor protects endothelial cells through the phosphoinositide 3-kinase–Akt–Bcl-2 pathway. J Biol Chem 279(41):43352–43360. doi:10.1074/jbc.M405777200
Liu RL, Xiong QJ, Shu Q, Wu WN, Cheng J, Fu H, Wang F, Chen JG, Hu ZL (2012) Hyperoside protects cortical neurons from oxygen-glucose deprivation-reperfusion induced injury via nitric oxide signal pathway. Brain Res 1469:164–173. doi:10.1016/j.brainres.2012.06.044
Pap M, Cooper GM (1998) Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. J Biol Chem 273(32):19929–19932
Gim SA, Sung JH, Shah FA, Kim MO, Koh PO (2013) Ferulic acid regulates the AKT/GSK-3β/CRMP-2 signaling pathway in a middle cerebral artery occlusion animal model. Lab Anim Res 29(2):63–69. doi:10.5625/lar.2013.29.2.63
Zhu L, Fratiglioni L, Guo Z, Aguero-Torres H, Winblad B, Viitanen M (1998) Association of stroke with dementia, cognitive impairment, and functional disability in the very old: a population-based study. Stroke 29(10):2094–2099
Prencipe M, Ferretti C, Casini AR, Santini M, Giubilei F, Culasso F (1997) Stroke, disability, and dementia: results of a population survey. Stroke 28(3):531–536
Shen H, Wu X, Zhu Y, Sun H (2013) Intravenous administration of achyranthes bidentata polypeptides supports recovery from experimental ischemic stroke in vivo. PLoS ONE 8(2):e57055. doi:10.1371/journal.pone.0057055
Zhang X, Yeung PK, McAlonan GM, Chung SS, Chung SK (2013) Transgenic mice over-expressing endothelial endothelin-1 show cognitive deficit with blood–brain barrier breakdown after transient ischemia with long-term reperfusion. Neurobiol Learn Mem 101:46–54. doi:10.1016/j.nlm.2013.01.002
Roof RL, Schielke GP, Ren X, Hall ED (2001) A comparison of long-term functional outcome after 2 middle cerebral artery occlusion models in rats. Stroke 32(11):2648–2657
Tatemichi TK, Foulkes MA, Mohr JP, Hewitt JR, Hier DB, Price TR, Wolf PA (1990) Dementia in stroke survivors in the Stroke Data Bank cohort. Prevalence, incidence, risk factors, and computed tomographic findings. Stroke 21(6):858–866
Dong DW, Zhang YS, Yang WY, Wang-Qin RQ, Xu AD, Ruan YW (2013) Hyperphosphorylation of tau protein in the ipsilateral thalamus after focal cortical infarction in rats. Brain Res. doi:10.1016/j.brainres.2013.11.004
Wada A, Yokoo H, Yanagita T, Kobayashi H (2005) Lithium: potential therapeutics against acute brain injuries and chronic neurodegenerative diseases. J Pharmacol Sci 99(4):307–321
Shi C, Zheng DD, Fang L, Wu F, Kwong WH, Xu J (2012) Ginsenoside Rg1 promotes nonamyloidgenic cleavage of APP via estrogen receptor signaling to MAPK/ERK and PI3K/Akt. Biochim Biophys Acta 1820(4):453–460. doi:10.1016/j.bbagen.2011.12.005
Hwang YP, Jeong HG (2010) Ginsenoside Rb1 protects against 6-hydroxydopamine-induced oxidative stress by increasing heme oxygenase-1 expression through an estrogen receptor-related PI3K/Akt/Nrf2-dependent pathway in human dopaminergic cells. Toxicol Appl Pharmacol 242(1):18–28. doi:10.1016/j.taap.2009.09.009
Zhang X, Shi M, Bjoras M, Wang W, Zhang G, Han J, Liu Z, Zhang Y, Wang B, Chen J, Zhu Y, Xiong L, Zhao G (2013) Ginsenoside Rd promotes glutamate clearance by up-regulating glial glutamate transporter GLT-1 via PI3K/AKT and ERK1/2 pathways. Front Pharmacol 4:152. doi:10.3389/fphar.2013.00152
Acknowledgments
The authors are grateful to Ms. Dongyun Feng for technical assistance. This study was supported by the National Natural Science Foundation of China (Nos. 81171236, 81371365, 31170801, and 31300900) and Program for Changjiang Scholars and Innovative Research Team in University (No. IRT1053).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhang, X., Shi, M., Ye, R. et al. Ginsenoside Rd Attenuates Tau Protein Phosphorylation Via the PI3K/AKT/GSK-3β Pathway After Transient Forebrain Ischemia. Neurochem Res 39, 1363–1373 (2014). https://doi.org/10.1007/s11064-014-1321-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-014-1321-3