
Abstract
Cerebral ischemia/reperfusion injury plays an important role in the development of tissue injury after acute stroke, including neutrophils adhesion and infiltration, inflammation and oxidative stress. 10-O-(N,N-dimethylaminoethyl)-ginkgolide B methanesulfonate (XQ-1H) is a novel ginkdolide B derivative. In this study, we investigated the anti-inflammatory and anti-oxidative activities of XQ-1H in vivo and vitro. In our study, rats were treating with XQ-1H (31.2, 15.6 and 7.8 mg/kg) after middle cerebral artery occlusion surgery. Primary cultured cortical rat neurons were treated with Na2S2O4 for 1.5 h to mimic hypoxia and reoxygenation injury in vitro. Cortical neurons were preincubated with XQ-1H (100, 10, 1 μM) 24 h before hypoxic injury. Brain edema was evaluated by brain water content. Neutrophil infiltration was determined by fluorescence imaging method and myeloperoxidase assay. Intercellular adhesion molecule 1 (ICAM-1) and matrix metallopeptidase 9 (MMP-9) expressions were examined by immunohistochemistry analysis. Neuronal injury was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide, lactate dehydrogenase releasing and lactic acid content. The anti-oxidative effects of XQ-1H were evaluated by superoxide dismutase (SOD) activity and malondialdehyde content in ischemic brain and neuron cultures subjected to hypoxia/reoxygenation procedure. Results showed that XQ-1H reduced neutrophils infiltration to ischemic brain, which might result from down regulation of inflammatory mediators, such as ICAM-1 and MMP-9. In addition, an antioxidative effect of XQ-1H was observed in cortical neuron and brain homogenates by enhancing SOD activity and inhibiting lipid peroxidation. These results indicated that XQ-1H possessed a protective effect against cerebral ischemia, especially on neutrophil infiltration and oxidative stress.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ischemia reperfusion can set off numerous cascades of secondary injury, especially many of inflammatory mediators and cells. Inflammation plays an important role in the pathogenesis of stroke, especially in the context of reperfusion. Reactive radicals will be generated, and blood brain barrier integrity may be compromised [1].
One of the manifestations of central nervous system damage caused by cerebral ischemia is the brain edema, which is a result of the breakdown of blood brain barrier (BBB) [2]. During cerebral ischemia, numbers of factors may result in the breakdown of BBB, in particular inflammation and oxidative stress.
After the interruption of cerebral blood flow (CBF), tissue injury begins with an inflammatory reaction, which requires the recruitment and infiltration of leukocytes, both polymorphonuclear neutrophils (PMNs) and monocytes/macrophages (but not lymphocytes). The rolling interaction appears to be largely mediated by P-selectin expressed on endothelial cells [3], while firm adhesion is mediated by an interaction between b2-integrins on leukocytes with ICAM-1 on cerebral microvascular endothelial cells [4]. Leukocytes adhere to the microvascular endothelial cell in the presence of activated PMNs, which eventually leads to the microvessel obstruction, edema formation, cellular necrosis, and tissue infarction [5, 6]. Besides, activation of PMNs causes the release of enzymes within cytoplasmatic vesicles [7]. Studies shown that myeloperoxidase (MPO) is one of the principal enzymes released from the neutrophil’s azurophilic granules [8]. MPO activity in brain tissue was evaluated as an index of neutrophil accumulation [9]. Numerous studies shown that neutrophil depletion alleviated cerebral tissue injury in the permanent or transient focal ischemia model in mice and rats [10].
One of the most well-studied mechanisms for BBB breakdown from oxidative stress is via matrix metalloproteinase (MMP) activation. Free radicals activate MMPs,which eventually enhances the extracellular matrix degradation activity and leads to the disruption of BBB via tight junction [11]. Recent research has indicated that MMP-9 protein and mRNA were significantly correlated with brain water content (BWC), which correlates with cerebral edema [12].
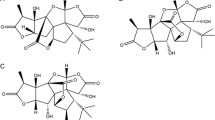
XQ-1H is a novel ginkgolide B derivative with the same active structure as ginkgolide B [13] (Fig. 1). A modification on dimethylamino-ethoxy group by combing with methane sulfonic acid helps to increase water solubility of XQ-1H for injection in clinical application. Previous studies have indicated that XQ-1H exhibits neuroprotective effect both in vitro and in vivo [14, 15].

Chemical structure of XQ-1H, 10-O-(N,N-dimethylaminoethyl)-ginkgolide B methanesulfonate
In this study, we investigated the effects of XQ-1H on neutrophils infiltration and oxidative stress in ischemic rats and cortical neurons subjected to hypoxia/reoxygenation injury.
Methods
Chemicals and Reagents
XQ-1H was kindly provided by Jiangsu Kefeiping Pharmaceutical Company Limited. Rhodamine -6G was purchased from Sigma. MPO, superoxide dismutase (SOD), malondialdehyde (MDA), lactate dehydrogenase (LDH) and lactic acid (LD) kits were purchased from Nanjing Jiancheng Bioengineering Institute. Minimum Essential Medium (MEM), Neurobasal medium (without Phenol Red and estrogen-free) and B-27 were purchased from Gibco. All other reagents were of analytical grade and commercially available.
Animal Care
Male Sprague-Dawley rats (250–300 g body weight, Zhejiang Laboratory Animals Center, Zhejiang Academy of Medical Science, Hangzhou, China) were used for the current study. All animal—use procedures were in strict accordance with institutional guidelines of China Pharmaceutical University (Nanjing, China). All experiments were approved by Animal Ethics Committee of China Pharmaceutical University.
Animal Preparation and Treatment
Ischemia and Reperfusion (I/R) Model
Animals were fasted with free access to water the night before surgery. All animals were randomly divided into 5 groups (n=8 for each group): sham, MCAO/R, XQ-1H (31.2 mg/kg), XQ-1H (15.6 mg/kg), XQ-1H (7.8 mg/kg). Rats were anesthetized with an intraperitoneal injection of 3% chloral hydrate (300 mg/kg). The local CBF of the middle cerebral artery territory was monitored during the entire duration of the experiment by laser—Doppler fluxmetry (MP150 Starter system; BIOPAC System, Inc., USA) [16]. Apart from sham group, all other groups were performed MCAO surgery [17]. The MCAO procedures have been developed to obtain a relatively low mortality (less than 40 %). The middle cerebral artery (MCA) was occluded using a silicone-coated nylon monofilament (diameter about 0.38 mm) inserted via the external carotid artery. Sufficient occlusion of the middle cerebral artery was monitored by laser -Doppler fluxmetry (the local cerebral blood flow was decreased to about 10–30 % of baseline). XQ-1H was dissolved in saline and injected via femoral vein after MCAO surgery. Sham group and MCAO/R group (vehicle group) were given an equivalent volume of saline. Reperfusion was initiated by withdrawing the filament after 2 h.
Measurement of Brain Water Content
To evaluate the BWC, the right hemisphere was superficially dried, and transferred to aluminum foil, weighed (wet weight), and dried overnight at 105 °C in a desiccating oven. The dried hemisphere was weighed again (dry weight) and the total brain water was calculated according to the following formula: [(wet weight−dry weight)/wet weight] × 100% [18].
Fluorescence Imaging on Neutrophils Infiltration After Focal Ischemia
24 h after MCAO, neutrophilic granulocytes were monitored by intravenous administration of 50 μl of 0.5 % rhodamine-6G before all animals sacrificed. 15 min after administration, brains were quickly removed and placed in Carestream In Vivo Imaging System, followed by a fluorescence imaging scanning (absorption peak of 525 nm; emission peak of 555 nm). Post processing of images taken by optical zoom lens makes it possible to depict and quantitate the obtained signals. The signals were analyzed and given as mean intensity respectively in right hemisphere and left hemisphere. The whole brain mean intensity (R/L) was calculated according to the following formula: right hemisphere mean intensity / left hemisphere mean intensity. Data were analyzed with Kodak Molecular Imaging Software 5.X.
Measurement of MPO, SOD and MDA in Brain Homogenates
24 h after MCAO, rats were sacrificed. Ischemic hemispheres were rapidly removed and measured the wet weight. Each sample was homogenized in ice phosphate buffer saline (1/9, wt./vol.). Supernatant was collected and detected for MPO, SOD activity and MDA content, using commercially available kits (Jiancheng Bioengineering Institute, Nanjing, China).
Immunohistochemistry Analysis of ICAM-1 and MMP-9 Expression in Ischemic Brain
The ICAM-1 and MMP-9 expressions in rats’ brain were detected using the immunohistochemistry (IHC) analysis [19]. Rats were anesthetized at 24 h after MCAO. The chest was subsequently opened and perfused with 4 % paraformaldehyde through the left ventricle until perfusion fluid obtained from the right atrium was colorless. Brains were rapidly removed. The right hemisphere tissue was fixated in 4% paraformaldehyde. Then the tissue samples were embedded in paraffin before sectioning at 4 μm. The paraffin sections were performed antigen retrieval and blocked in normal goat serum for 10 min. Then sections were incubated with primary antibody at 37 °C for 1 h after serum discarded. All sections were washed three times with PBS. Sections were then incubated with secondary antibody (MaxVision™ IHC Kit) at 37 °C for 15 min, washed with PBS, and incubated with DAB substrate for 3-5 min. All antibodies, were dissolved in PBS (containing 1% v/v normal goat serum and 0.3% v/v Triton X-100, pH 7.4).Controls for the immunohistochemistry procedure were routinely performed without incubation with primary antibody. The control brain sections did not develop any immunohistochemical labeling. Light microscope images were taken in a LEICA DM 1,000 microscope. The amount of IHC dye is linearly related to the optical density (absorbance). Optical density of staining was obtained from light microscope images using MiniSee software. The expressions of ICAM-1 and MMP-9 were finally given as mean optical density value.
Cell Cultures Preparation and Treatment
Primary Rats Neuron Cultures
Rats’ cortical neurons were prepared essentially as described [20], with minor modifications. 0.02 % ethylene diamine tetraacetic acid (EDTA) solution and 0.25 % trypsin solution were needed to prepare cell dissociation solution. Cortices were obtained from embryonic day 18 (E18) Sprague-Dawley rat embryos previously, triturated in ice PBS for 1 min (90 % tissue dispersal). Tissue dispersal was incubated in sterile tube with 2 ml of cell dissociation solution at 37 °C for 10 min. The dissociation was terminated with Minimum Essential Medium (MEM) containing 10 % fetal serum. The dissociated samples were filtered using 35 μm nylon mesh. The suspension was centrifuged at 1,000 rpm for 5 min, Then cells were resuspended in MEM supplemented with glucose (0.6 % wt./vol.), penicillin (100 U/ml), streptomycin (100 mg/ml), and 10 % fetal calf serum. Cells were diluted with MEM to approximately 1 × 109/L and plated into 6-well-plates (or 24-well-plates), which were previously coated with 10 mg/l poly-l-lysine. Cells were incubated at 37 °C, 5 % CO2, 95 % humidity for 6 h. After neurons were attached to the plate, the medium was discarded, and neurons were maintained in Neurobasal medium supplemented with 2 % B-27 and l-glutamine (0.5 mM).
Hypoxia/Reoxygenation Model and Cell Culture Treatment
The cells were treated with Na2S2O4 at concentration of 20 mM in the glucose-free Earle’s balanced salt solution (EBSS, pH 7.4) medium for 1.5 h (hypoxia). Hypoxia/reoxygenation procedure was terminated by replacing the anoxic medium with Neurobasal medium for an additional 2 h (reoxygenation). Various concentrations of XQ-1H (100, 10, 1 μM) were added into the cultures 24 h before inducing hypoxia/reoxygenation. Control cultures were treated in an identical way without inducing hypoxic injury.
MTT Assay
The protective effects of XQ-1H on cortical neurons were measured by quantitative colorimetric assay with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) with minor modifications, showing the mitochondrial activity of living cells. Neurons were exposed or not exposed to Na2S2O4 induced hypoxia/reoxygenation before MTT assay. MTT was added to 24-well- plate at a final concentration of 0.5 mg/ml for 4 h at 37 °C, and the insoluble formazan crystals were dissolved in 200 μl of DMSO. Cells viability was quantified and given as the optical density value read at 570 nm.
Measurement of SOD, MDA, LDH and LD in Neuron Cultures
Neuronal injury was quantitatively assessed by the measurement of LDH releasing into the culture medium and intracellular SOD, MDA and LD after OGD injury, using commercially available kits.
Statistics
All experimental results are given as mean ± S.D., and statistical significance was analyzed by one-way analysis of variance (ANOVA) test. Results were considered to be statistically significant when P < 0.05.
Results
XQ-1H Alleviated Brain Edema After Ischemia/Reperfusion
To measure brain edema formation, we measured the BWC (%). Results (shown in Fig. 2) revealed that the BWC in MCAO/R group documented significant edema formation after cerebral ischemia (82.3 ± 1.7 %). Compared to MCAO/R group, edema formation was alleviated by injecting XQ-1H (31.2 mg/kg) after MCAO surgery (P < 0.01).

Effects of XQ-1H on brain water content in MCAO rats. Values are mean ± S.D. of 8 samples for each group. △△ P<0.01 versus sham group value, *P < 0.05, ** P < 0.01 versus MCAO/R group value
XQ-1H Suppressed Neutrophils Infiltration to Brain Parenchyma
The time point 24 h after ischemia-reperfusion was chosen because previous studies suggest neutrophils infiltration into the ischemic region of the brain occurs early after ischemia-reperfusion. An area of fluorescence was significantly observed in the right hemisphere of the brain (the left hemisphere in the image) after the onset of ischemia, as shown in MCAO/R group, which indicated labeled neutrophils infiltrations into the brain tissue. The graphs and results (shown in Fig. 3a) suggested that neutrophils infiltration was alleviated by treatment with XQ-1H at a dose-dependent manner, which compared to MCAO/R group (P < 0.01, P < 0.05).

Protective effects of XQ-1H on neutrophils infiltration after ischemia reperfusion. Effects of XQ-1H on neutrophil infiltration in ischemic brain (a). Effects of XQ-1H on MPO activity in brain homogenates (b). Values are mean ± S.D. of 8 samples for each group. △△ P<0.01 versus sham group value, *P < 0.05, **P < 0.01 versus MCAO/R group value
In our study, we certified neutrophil infiltration by measuring MPO activity. The results (shown in Fig. 3b) revealed that treatment with XQ-1H (31.2 and 15.6 mg/kg, i.v) may significantly reduce MPO activity compared to the MCAO/R group (P < 0.01, P < 0.05).
XQ-1H Downregulated the Expression of ICAM-1 and MMP-9 After Ischemia-Reperfusion Injury
To investigate the protective role of XQ-1H in blood brain barrier, we examined the expression of ICAM-1 and MMP-9 in ischemic brain. Our results (shown in Fig. 4) showed that cerebral ischemia may lead to over-expression of ICAM-1 and MMP-9, which was in accordance with previous studies. The over-expression of two mediators was significantly attenuated by XQ-IH.

Effects of XQ-1H on ICAM-1 and MMP-9 expression after ischemia reperfusion. Values are mean ± S.D. of 8 samples for each group. ΔΔ P < 0.01 versus sham group value, *P < 0.05, **P < 0.01 versus MCAO/R group value, # P < 0.05, ## P < 0.01 versus MCAO/R group value
XQ-1H Protected Cortical Neurons Against Hypoxic Injury
Hypoxia/reoxygenation elicited remarkable neurons injury. These results were demonstrated by MTT assay, LDH releasing and LD accumulation assay. MTT assay showed that neurons viability after hypoxic injury significantly decreased compared with control group. Pretreatment with XQ-1H at different concentrations significantly reversed neuronal injury (shown in Fig. 5a). 100 μM XQ-1H exhibited the maximal neuron protection effect, which was also certified by LDH releasing and LD accumulation assay. LDH and LD assay demonstrated that XQ-1H improved neuronal energy by significantly reducing LDH releasing and LD accumulation (shown in Fig. 5b, c). Moreover, the toxicity study indicated XQ-1H did not affect cell viability under normal condition at these three concentrations (shown in Fig. 5a).

Protective effects of XQ-1H on hypoxia/reoxygenation induced cortical neuron injury. Effects of XQ-1H on neuronal viability after cultures exposed to hypoxia/reoxygenation or not exposed to hypoxia/reoxygenation (a). Effects of XQ-1H on LDH release after hypoxia/reoxygenation (b). Effects of XQ-1H on LD accumulation after hypoxia/reoxygenation (c). Values are mean ± S.D. of 6 samples for each group. △△ P<0.01 versus control group value, *P < 0.05, **P < 0.01 versus vehicle group value
XQ-1H Exhibited Cerebral Protection Effects Against Oxidative Stress In Vitro and Vivo
Results (Table 1) suggested that SOD activity significantly enhanced in brain homogenates by treatment with XQ-1H at a dose-dependent manner. In our study, increased SOD activity in ischemic brain indicated that XQ-1H possessed antioxidant property (31.2, 15.6 mg/kg), which was certified by MDA assay (Table 1). We also evaluated neuronal damage by measuring SOD activity and MDA content (Table 2). Similar effects were observed in XQ-1H treated neurons. Compared to vehicle cultures, XQ-1H significantly enhanced cellular antioxidant defenses, with 50.5 ± 6.1, 62.4 ± 4.1 and 68.4 ± 9.6 of SOD activity (Table 2). This might lead to the inhibition of lipid peroxidation, with 4.56 ± 1.47, 3.93 ± 0.74, 2.77 ± 0.51 of MDA production (Table 2).
Discussion
This study aimed to investigate the protective role of XQ-1H against neutrophil infiltration and oxidative stress after ischemia reperfusion. Our results demonstrated that XQ-1H exhibited protective effect against ischemia reperfusion insult in vivo by alleviating neutrophils infiltration and inflammatory mediators to the brain parenchyma, such as ICAM-1 and MMP-9. Besides, extenuated oxidative stress injury was observed after XQ-1H treatment through enhancing SOD and inhibiting MDA activity.
Leukocyte recruitment persists for days to weeks following the ischemic insult, population of recruited cells shifts from PMNs to mononuclear leukocytes [21]. Furthermore, increased neutrophils adhesion has been demonstrated to be one of the most important factors in the pathogenesis of inflammatory injury during I/R injury of the brain [22]. In our study, rhodamine 6G was used for labeling neutrophils in the brain. Our present study represented the first attempt to evaluate the effect of XQ-1H against neutrophils infiltration on transient ischemia and reperfusion. We found that 2 h of MCAO,followed by 22 h of reperfusion significantly elicited neutrophils infiltration within brain tissue. Three doses of XQ-1H significantly decreased neutrophils infiltration in transient MCAO experimental animal, which was also certified by MPO concentration in brain tissue. Previous studies have shown that MPO activity in brain tissue was determined as an index of neutrophil accumulation [7, 23, 24]. Down regulation of ICAM-1 expression by treatment of XQ-1H implicated that XQ-1H alleviated neutrophils infiltration through suppressing firm adhesion of PMN to endothelial cells. However, whether XQ-1H inhibited neutrophils infiltration through suppressing neutrophils rolling remains unclear. Further work will be required to elucidate the precise molecular mechanisms involved.
An important source of MMP-9 is from perivascular neutrophils [25]. Cerebrovascular basal lamina and its tight junctions comprise of numerous molecular constituents, which are substrates for activated MMP-9 [26, 27]. A degradation of a basic BBB component such as collagen IV occurs in microvasculars presentating an important infiltration of MMP-9+ neutrophils [25]. In our study, we also examined the protective effect of XQ-1H on BBB, including MMP-9 expression and BWC measurement. Results showed that MMP-9 expression in ischemic brain was down-regulated after XQ-1H administration, followed by an alleviation in brain edema, which indicated that XQ-1H prevented BBB disruption.
Inflammation is generally associated with the enhanced production of reactive oxygen species (ROS). In the acute phase (minutes to hours) of ischemic stroke, ROS and proinflammatory mediators (cytokines and chemokines) are released rapidly from injured tissue [28, 29]. These mediators induce the expression of the adhesion molecules on both cerebral endothelial cells and leukocytes, eventually promote the adhesion and transendothelial migration of circulating leukocytes [30]. In the subacute phase (hours to days), infiltrating leukocytes release cytokines and chemokines, such as excessive production of ROS and activated MMPs (mainly MMP-9), which eventually amplify the brain-inflammatory responses further by causing more extensive activation of resident cells and infiltration of leukocytes, eventually leading to disruption of the BBB, brain edema, neuronal death, and hemorrhagic transformation [28, 29]. In a previous study, infarct size and neurological deficits were measured and presented by Sun J et al. [31]. The results indicated that XQ-1H would potentially improve neurological injury. On the other hand, an antioxidative effect of XQ-1H was observed in our study, but whether this mechanism contributed to the neutrophils infiltration still need further investigation.
In conclusion, less neutrophils infiltration was observed in ischemic brain by treatment with XQ-1H, which might result from down regulation of inflammatory mediators, like ICAM-1 and MMP-9. Our teams still work on the mechanism of XQ-1H on oxidative stress, and hope to explain the relationship between oxidative stress and neutrophil infiltration in ischemia.
References
Xing C, Arai K, Lo EH, Hommel M (2012) Pathophysiologic cascades in ischemic stroke. Int J Stroke 7(5):378–385
Kamada H, Yu F, Nito C, Chan PH (2007) Influence of hyperglycemia on oxidative stress and matrix metalloproteinase-9 activation after focal cerebral ischemia/reperfusion in Rats: relation to blood-brain barrier dysfunction. Stroke 38(3):1044–1049
Ishikawa M, Cooper D, Russell J, Salter JW, Zhang JH, Nanda A, Granger DN (2003) Molecular determinants of the prothrombogenic and inflammatory phenotype assumed by the postischemic cerebral microcirculation. Stroke 34(7):1777–1782
Liu L, Kubes P (2003) Molecular mechanisms of leukocyte recruitment: organ-specific mechanisms of action. Thromb Haemost 89(2):213–220
Chou WH, Choi DS, Zhang H, Mu D, McMahon T, Kharazia VN, Lowell CA, Ferriero DM, Messing RO (2004) Neutrophil protein kinase Cdelta as a mediator of stroke reperfusion injury. J Clin Invest 114(1):49–56
Clark RK, Lee EV, Fish CJ, White RF, Price WJ, Jonak ZL, Feuerstein GZ, Barone FC (1993) Development of tissue damage, inflammation and resolution following stroke: an immunohistochemical and quantitative planimetric study. Brain Res Bull 31(5):565–572
Jordan JE, Zhao ZQ, Vinten-Johansen J (1999) The role of neutrophils in myocardial ischemia-reperfusion injury. Cardiovasc Res 43(4):860–878
Klebanoff SJ (1999) Myeloperoxidase. Proc Assoc Am Physicians 111(5):383–389
Bradley PP, Christensen RD, Rothstein G (1982) Cellular and extracellular myeloperoxidase in pyogenic inflammation. Blood 60(3):618–622
Jean WC, Spellman SR, Nussbaum ES, Low WC (1998) Reperfusion injury after focal cerebral ischemia: the role of inflammation and the therapeutic horizon. Neurosurgery 43(6):1382–1397
Gu Y, Zheng G, Xu M, Li Y, Chen X, Zhu W, Tong Y, Chung SK, Liu KJ, Shen J (2012) Caveolin-1 regulates nitric oxide-mediated matrix metalloproteinases activity and blood-brain barrier permeability in focal cerebral ischemia and reperfusion injury. J Neurochem 120(1):147–156
Li DD, Song JN, Huang H, Guo XY, An JY, Zhang M, Li Y, Sun P, Pang HG, Zhao YL, Wang JF (2013) The roles of MMP-9/TIMP-1 in cerebral edema following experimental acute cerebral infarction in rats. Neurosci Lett 550:168–172
Liu W, Li P, Feng F, Mao L (2010) Isolation and structure characterization of related impurities in 10-O-(N,N-dimethylaminoethyl)-ginkgolide B methanesulfonate (XQ-1H) bulk drug and quantitation by a validated RP-LC. J Pharm Biomed Anal 52(4):603–608
Deng Y, Fang W, Li Y, Cen J, Fang F, Lv P, Gong S, Mao L (2009) Blood-brain barrier breakdown by PAF and protection by XQ-1H due to antagonism of PAF effects. Eur J Pharmacol 616(1–3):43–47
Yang Q, Fang W, Lv P, Geng X, Li Y, Sha L (2012) Therapeutic neuroprotective effects of XQ-1H in a rat model of permanent focal cerebral ischemia. Pharmacology 89(1–2):1–6
Gamboa J, Blankenship DA, Niemi JP, Landreth GE, Karl M, Hilow E, Sundararajan S (2010) Extension of the neuroprotective time window for thiazolidinediones in ischemic stroke is dependent on time of reperfusion. Neuroscience 170(3):846–857
Longa EZ, Weinstein PR, Carlson S, Cummins R (1989) Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20(1):84–91
Mdzinarishvili A, Kiewert C, Kumar V, Hillert M, Klein J (2007) Bilobalide prevents ischemia-induced edema formation in vitro and in vivo. Neuroscience 144(1):217–222
Lazarowski A, Caltana L, Merelli A, Rubio MD, Ramos AJ, Brusco A (2007) Neuronal mdr-1 gene expression after experimental focal hypoxia: a new obstacle for neuroprotection? J Neurol Sci 258(1–2):84–92
Qian Y, Guan T, Tang X, Huang L, Huang M, Li Y, Sun H (2011) Maslinic acid, a natural triterpenoid compound from Olea europaea, protects cortical neurons against oxygen-glucose deprivation-induced injury. Eur J Pharmacol 670(1):148–153
Barone FC, Hillegass LM, Tzimas MN, Schmidt DB, Foley JJ, White RF, Price WJ, Feuerstein GZ, Clark RK, Griswold DE et al (1995) Time-related changes in myeloperoxidase activity and leukotriene B4 receptor binding reflect leukocyte influx in cerebral focal stroke. Mol Chem Neuropathol 24(1):13–30
Jin R, Yang G, Li G (2010) Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol 87(5):779–789
Zhang RL, Chopp M, Jiang N, Tang WX, Prostak J, Manning AM, Anderson DC (1995) Anti-intercellular adhesion molecule-1 antibody reduces ischemic cell damage after transient but not permanent middle cerebral artery occlusion in the Wistar rat. Stroke 26(8):1438–1442
Zhang ZG, Chopp M, Tang WX, Jiang N, Zhang RL (1995) Postischemic treatment (2–4 h) with anti-CD11b and anti-CD18 monoclonal antibodies are neuroprotective after transient (2 h) focal cerebral ischemia in the rat. Brain Res 698(1–2):79–85
Rosell A, Cuadrado E, Ortega-Aznar A, Hernández-Guillamon M, Lo EH, Montaner J (2008) MMP-9-positive neutrophil infiltration is associated to blood-brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke 39(4):1121–1126
Tilling T, Korte D, Hoheisel D, Galla HJ (1998) Basement membrane proteins influence brain capillary endothelial barrier function in vitro. J Neurochem 71(3):1151–1157
Van den Steen PE, Dubois B, Nelissen I, Rudd PM, Dwek RA, Opdenakker G (2002) Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9). Crit Rev Biochem Mol Biol 37(6):375–536
Amantea D, Nappi G, Bernardi G, Bagetta G, Corasaniti MT (2009) Post-ischemic brain damage: pathophysiology and role of inflammatory mediators. FEBS J 276(1):13–26
Kriz J (2006) Inflammation in ischemic brain injury: timing is important. Crit Rev Neurobiol 18(1–2):145–157
Yilmaz G, Granger DN (2008) Cell adhesion molecules and ischemic stroke. Neurol Res 30(8):783–793
Sun J, Li Y, Fang W, Mao L (2011) Therapeutic time window for treatment of focal cerebral ischemia reperfusion injury with XQ-1 h in rats. Eur J Pharmacol 666(1–3):105–110
Acknowledgments
This study was supported by Natural Science Foundation of Jiangsu Province of China (Program No. BK2011625), National Natural Science Foundation of China (Program No. 81202974), Project Program of State Key Laboratory of Natural Medicines, China Pharmaceutical University (No. JKGQ201108), and sponsored by Qing Lan Project.
Author information
Authors and Affiliations
Corresponding author
Additional information
Jie Wei and Weirong Fang have contributed equally to this work.
Rights and permissions
About this article
Cite this article
Wei, J., Fang, W., Sha, L. et al. XQ-1H Suppresses Neutrophils Infiltration and Oxidative Stress Induced by Cerebral Ischemia Injury Both In Vivo and In Vitro. Neurochem Res 38, 2542–2549 (2013). https://doi.org/10.1007/s11064-013-1176-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-013-1176-z