
Abstract
Resveratrol has several beneficial effects, including reductions of oxidative stress, inflammatory responses and apoptosis. It has been known that resveratrol is a sirtuin 1 (SIRT1) activator and protective effects of resveratrol are mediated by Akt and mitogen-activated protein kinases. However, it is not examined whether these pathways are regulated by resveratrol in the ischemic brain. Previously, we found that acute resveratrol treatment reduces brain injury induced by transient focal ischemic stroke. In the present study, we defined the signaling pathways modulated by resveratrol in ischemia by examining SIRT1 expression and phosphorylation of Akt, ERK1/2 and p38 in the ischemic cortex. Resveratrol increased expression of SIRT1 and phosphorylation of Akt and p38 but inhibited the increase in phosphorylation of ERK1/2. Gene and protein levels of peroxisome proliferator-activated receptor γ coactivator 1α, a downstream molecule of SIRT1, and mRNA levels of its target genes antioxidative superoxide dismutase 2 and uncoupling protein 2 were elevated. Resveratrol also increased phosphorylation of cyclic AMP-response-element-binding protein and transcription of the anti-apoptotic gene Bcl-2. These results suggest that various neuroprotective actions of resveratrol, including anti-oxidative, anti-apoptotic and inflammatory effects, are mediated via modulation of multiple signaling pathways in the ischemic brain.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Resveratrol, a polyphenol found in red wine, is considered as a potential drug in the treatment of cardiovascular disease, diabetes, cancer, and neurological diseases, including ischemic brain disease [1, 2]. When administered prior to or following ischemia in rodent models, resveratrol reduces the size of brain lesions [3–9]. The neuroprotective mechanisms of resveratrol may include anti-oxidation [6–9], anti-inflammation [3, 9], and anti-apoptosis [10]. Given these diverse actions, it is likely that multiple signaling pathways are involved in the neuroprotection afforded by resveratrol.
Sirtuin 1 (SIRT1) is a nicotine adenine dinucleotide (NAD+)-dependent deacetylase that regulates various proteins involved in biological processes [11]. Resveratrol is known as a SIRT1 activator, and therefore it has been shown that SIRT1 activity is necessary for resveratrol-mediated neuroptrotection in models of neurological disorders [12–16]. Peroxisome proliferator-activated receptor-gamma coactivator 1α (PGC1α), a master regulator of anti-oxidative enzymes and mitochondrial biogenesis [17], has been identified as a target protein of SIRT1 in models of global ischemia, amyotrophic lateral sclerosis and Parkinson’s disease [12, 14, 15]. Mitochondrial antioxidant enzymes are induced by PGC1α in neurons exposed to oxidative stress, indicating that increasing PGC1α expression protects neurons from oxidative insult [17]. A study using a focal ischemic stroke model showed that increased SIRT1-dependent PGC1α expression played a crucial role in neuroprotection induced by a flavonoid icariin [18]. Therefore, it is possible that SIRT1/PGC1α signaling contributes to resveratrol-mediated anti-oxidative effects in the ischemic brain.
The serine/threonine kinase Akt (a downstream effector of phosphatidylinositol 3-kinase) and mitogen-activated protein kinases (MAPKs; e.g., extracellular signal-regulated kinase1/2 [ERK1/2] and p38) are important determinants of the outcome of ischemic stroke through their regulation of multiple signals [19, 20]. Activation of Akt is associated with reduction of ischemic brain damage via blockade of the apoptotic pathway [20]. The pro-survival action of Akt is mediated by a transcription factor, cyclic AMP response element-binding protein (CREB), which regulates transcription of the anti-apoptotic gene Bcl-2 in neurons [21–23]. It has been reported that resveratrol reduces ischemia-related cell death in the brain by reducing release of mitochondrial cytochrome c, which initiates apoptosis [10], but its signaling pathways have not been defined. Phosphorylated ERK1/2 and stress-activated protein kinase p38, which are induced by ischemic insults in the brain, are linked to cell survival or apoptosis [19, 24]. In addition, both ERK1/2 and p38 are involved in brain inflammatory responses by producing pro-inflammatory cytokines from activated microglia or astrocytes [25]. An anti-inflammatory effect of resveratrol has been shown in lipopolysaccaride-activated microglia or hippocampal slices exposed to glutamate through suppression of MAPKs [26–29]. Therefore, MAPK signaling pathways might be important for resveratrol-mediated neuroprotection in the ischemic brain.
Recently, we reported that a single treatment of resveratrol during acute periods after ischemic stroke in mice reduces infarct volume, and that resveratrol’s anti-oxidative and anti-inflammatory actions are associated with neuroprotection [9]. In the present study we examined whether SIRT1, Akt, ERK1/2, p38, and their downstream proteins in the ischemic brain are modulated and associated with the neuroprotective actions of resveratrol.
Materials and Methods
Transient Middle Cerebral Artery Occlusion (MCAO)
Male C57BL/6 mice, aged 10–11 weeks, were used in the experiments (Orient Bio Inc., Seongnam, Republic of Korea). All procedures were approved by the Institutional Animal Care and Use Committee at the Medical School of Ewha Womans University and conformed to international guidelines on the ethical use of animals. Mice were acclimatized for 1 week before the experiments in an animal room under a 12-h light/dark cycle at 22 ± 2 °C. The number of animals used for the study was minimized to reduce animal suffering.
Procedures for transient MCAO were described previously [9]. Briefly, mice were anesthetized with isoflurane, and a fiber optic probe was attached to the right parietal bone (2 mm posterior and 5 mm lateral to bregma) and connected to a laser-Doppler flowmeter (Periflux System 5010, Perimed, Sweden). Cerebral blood flow (CBF) was continuously recorded during MCAO and reperfusion periods with a computer-based data acquisition system (Perisoft, Perimed, Sweden). A 6–0 silicon-coated black monofilament surgical suture (Doccol Cooperation, Redlands, CA, USA) was inserted into the exposed right external carotid artery, advanced into the internal carotid artery, and wedged into the circle of Willis to obstruct the origin of the MCA. The filament was left in place for 30 min and then withdrawn to re-establish CBF. Only animals that exhibited a reduction in CBF >85 % during MCA occlusion and in which CBF recovered by >80 % after 10 min of reperfusion were included in the study. Rectal temperature was maintained at 37.0 ± 0.5 °C with a thermostatically controlled heating pad during surgery and recovery until the animal regained consciousness.
Drug Administration
Resveratrol (Sigma, St. Louis, MO, USA) was dissolved in ethanol and diluted in normal saline. The final concentration of ethanol in normal saline (vehicle) was 30 % (v/v) and total volume of vehicle administered to each mouse was 2 ml/kg. Mice were divided randomly into control and treatment groups, and vehicle or resveratrol (5 mg/kg) was injected into tail vein 3 h after MCAO as reported in the previous study [9].
Infarct Volume Measurement
Infarct volume was measured according to the procedures described previously [30]. Mice were sacrificed 3 days after MCAO; brains were removed, frozen and sectioned (30 μm thicknesses) using a cryostat. Brain sections were collected serially at 600-μm intervals, and stained with cresyl violet. Infarct volume was determined using an image analyzer (Axiovision LE 4.1, Carl Zeiss, Jena, Germany). Values were reported after correction for post-ischemic swelling as previously described [30].
RNA Isolation and Real-Time PCR Analysis
RNA was isolated from the ipsilateral hemisphere cortex and subjected to quantitative real-time PCR. Total RNA was prepared with Trizol reagent (Invitrogen Life Technologies, Carlsbad, CA, USA). Tissues were homogenized by sonicating in 0.5 ml Trizol, and RNA was purified using chloroform/isopropanol precipitation and dissolved in diethylpyrocarbonate-treated water (iNtRON Biotechnology, Seongnam, Republic of Korea). First strand synthesis was performed with 1 μg of total RNA from each sample and 1 μl (6.7 μM) of oligo (dT)15, with a Power cDNA Synthesis kit (iNtRON Biotechnology). A 2-μl aliquot of diluted cDNA (1:10) was amplified with SYBR® Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) in a final volume of 20 μl. PCR was performed in an ABI Prism 7000 sequence detector (Applied Biosystems). PCR cycles consisted of initial denaturation step at 95 °C for 5 min, followed by 40 cycles of 95 °C for 30 s, 54 °C for 30 s, and 72 °C for 45 s. The primers for actin (forward: 5′-AGGCTGTGCTGTCCCTGTAT-3′; reverse: 5′-AAGGAA GGCTGGAAAAGAGC-3′), PGC1α (forward: 5′-CTCACAGAGACACTGGACAGT-3′; reverse: 5′-TGT AGCTGAGCTGAGTGTTGG-3′), Bcl-2 (forward: 5′-CGCGTTGGCCCTTCGGAGTT-3′; reverse: 5′-ACA CTCCG CTTCACTGAGA-3′), UCP2 (forward: 5′-GCCCGGGCTGGTGGTGGTC-3′; reverse: 5′-CCC CGAAGGCAGAAGTGAAGTGG-3′) and sodium dioxide 2 (SOD2, forward: 5′-ATGTTGTGTCGGGC GCG-3′; reverse: 5′-AGGTAG TAAGCGTGCTCCCACACG-3′) were purchased from Bioneer (Daejoen, Republic of Korea). Cycling threshold (Ct) values of target genes were normalized to the Ct values of actin, and the relative expression level was calculated with the 2−ΔΔCt method [26]. All samples were run in triplicate, and naïve animals served as controls.
Western Blot Analysis
Cortical tissue from the ipsilateral hemisphere at 6 and 24 h after MCAO in each animal was used for western blotting. The tissue was lysed in 10 ml sodium dodecyl sulfate (SDS) buffer (62 mM Tris–HCl, 1 mM ethylenediamine tetraacetic acid, 2 % SDS, pH 6.8–7.0) containing a protease inhibitor cocktail (Complete Mini, Boehringer Mannheim, Germany), incubated for 20 min on ice, and centrifuged at 15,700×g for 10 min at 4 °C. Protein concentration of the supernatant was determined (Bio-Rad Laboratories, Hercules, CA, USA), and 80 μg of protein was loaded for SDS–polyacrylamide gel electrophoresis. Protein was transferred to Immobilon-P transfer membranes (Millipore Corporate, Billerica, MA, USA) using an electroblotting apparatus. Membranes were blocked for 1 h in Tris-buffered saline (TBS) containing 0.1 % Tween-20 and 5 % dry milk, incubated overnight with antibodies against SIRT1 (1:2,000; Millipore Corporate), Ser-473 pAkt (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA, USA), pERK1/2 (1:500; Santa Cruz Biotechnology), PGC1α (1:500; Santa Cruz Biotechnology), Tyr-182 phosphorylated p38 (pp38, 1:500; Santa Cruz Biotechnology), or Ser-133 pCREB (1:500; Millipore Corporate). Membranes then were washed three times (20 min each) with TBS containing 0.1 % Tween-20, incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h and washed three times (20 min each) with TBS containing 0.1 % Tween-20. Protein bands were visualized with the Western Blotting Luminal Reagent (Santa Cruz Biotechnology). Each membrane was re-probed with anti-actin (1:1,000), anti-Akt (1:1,000), anti-ERK1/2 (1:2,000), p38 (1:1,000) and anti-CREB (1:1,000; all from Santa Cruz biotechnology) antibodies after stripping with a stripping solution (Chemicon International, Temecula, CA, USA). For quantification, densities of the SIRT1 and PGC1α bands were normalized to the density of actin, and densities of the pAkt, pERK1/2, pp38 and pCREB bands were normalized to the density of the total (phosphorylated + unphosphorylated) forms of these proteins as measured with the Image J 1.37v program (NIH, Bethesda, MD, USA). Values are expressed as ratios versus controls that did not receive any surgical stimulus (given a nominal value of 1) at each time point after normalization.
Immunofluorescence Staining
Animals were anesthetized with sodium pentobarbital (120 mg/kg, i.p.) 24 h after MCAO, and perfused transcardially with saline followed by cold 4 % formaldehyde in 0.1 M sodium phosphate buffer (pH 7.2). The brains were removed, incubated overnight in fixative, and stored in a 30 % sucrose solution. Using a cryostat, serial coronal brain sections (20-μm thick, 600-μm intervals) were taken throughout the region spanning +1.4 to −1.0 mm from bregma and mounted on gelatin-coated slides. Brain sections were incubated in TBS containing 0.1 % Triton X-100, 5 % normal serum, and 1 % bovine serum albumin for 1 h, and then were incubated overnight in primary antibodies for pAkt (1:500), pCREB (1:500) and NeuN (1:1,000; Chemicon International). Secondary antibodies conjugated to Alexa fluor®568 (1:1,000; Molecular Probes, Eugene, OR, USA) and fluorescein isothiocyanate (1:1,000; Vector Laboratories, Inc., Burlingame, CA, USA) were applied to the sections for 1 h. Sections were washed with TBS between all steps. Vectashield mounting medium containing 6′-diamidino-2-phenylindole (DAPI, Vector Laboratories, Inc.) to label nuclei was applied to brain sections before placing the coverslip. Fluorescence images were obtained at a 4-μm thickness through the z axis of the section, covering a total of 20 μm in depth, using a confocal microscope (LSM5 PASCAL; Carl Zeiss) equipped with a filter set with excitation at 488 and 543 nm.
Statistical Analysis
Data are expressed as mean ± SEM. Comparisons between two groups were analyzed with an unpaired Student’s t test. Multiple comparisons were evaluated with one-way analysis of variance (ANOVA) followed by post hoc Fisher’s protected least significant difference (PLSD) tests, using the Statview software package (Statview version 5, SAS Institute Inc., Cary, NC, USA). Statistical significance was set at p < 0.05.
Results
Ischemic Cortex Was Rescued by Resveratrol
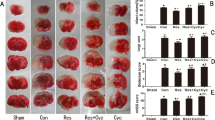
To verify protective effects of resveratrol against damage induced by ischemic stroke, infarct volumes were examined 3 days after MCAO. Total infarct volume was reduced significantly in mice treated with resveratrol as compared to mice treated with vehicle (45 % reduction; p < 0.01). The reduction was prominent in the cortex (36 % reduction; p < 0.01) but not in the striatum (p = 0.20) (Fig. 1a, b). CBF changes did not differ before vehicle or resveratrol treatment (Fig. 1c). These results confirm our previous finding of a therapeutic effect of acute resveratrol administration after stroke [9].

Acute resveratrol treatment reduced infarct volumes. a Representative brain sections of cresyl violet staining 3 days after MCAO in mice treated with vehicle or resveratrol. Dotted lines in brain sections indicated infracted areas. b Infarct volumes in total brain, cortex, and striatum 3 days after MCAO. Vehicle (n = 8) or resveratrol (5 mg/kg, n = 8) was administered into tail vein at 3 h after 30 min MCAO. c CBF changes during and 10 min after MCAO in mice treated with vehicle or resveratrol (n = 8 each). The data are presented as mean ± S.E.M. *p < 0.05 versus vehicle treatment (Student’s t test)
Expression Changes of SIRT1, pAKT, pERK1/2 and p38 Proteins by Resveratrol
To determine whether SIRT1, pAKT, pERK1/2 and pp38 were modulated by resveratrol, we analyzed protein levels of each protein at 6 and 24 h after MCAO. Because the reduction of infarct volume was significant in the cortex, we used cortical tissue to determine protein levels. At 6 h post-MCAO, SIRT1, pAkt and pp38 levels were increased (Fig. 2a, b and d) but pERK1/2 levels were unchanged (Fig. 2c) in both the vehicle and resveratrol-treated mice as compared to controls. There were no differences between vehicle and resveratrol treatments at the 6-h time point. At 24 h post-MCAO, the MCAO-induced increases in SIRT1 and pp38 expression were sustained in the vehicle-treated mice, but pAkt levels had normalized and were no longer different from those seen in controls (Fig. 2a, b and d). Resveratrol significantly increased expression of SIRT1 (1.36-fold change), pAkt (1.70-fold change), and pp38 (1.38-fold change) as compared to vehicle (all p’s < 0.05). By contrast, pERK1/2 levels at 24 h post-MCAO were markedly increased in the vehicle (1.82-fold change relative to control; p < 0.01) but not in the resveratrol-treated mice (0.66-fold change relative to vehicle; p < 0.01). Taken together, these results showed that resveratrol enhanced SIRT1 and pAkt expression, but differentially modulated activation of ERK1/2 and p38, 24 h after MCAO.

Resveratrol effects on the levels of SIRT1 (a), pAkt (b), pERK1/2 (c) and pp38 (d) in the cortex at 6 and 24 h after MCAO. Values are expressed as ratios versus normal controls (given a nominal value of 1) at each time point after normalization. SIRT1 was normalized to actin. Phosphorylated form of Akt, ERK1/2 and p38 were normalized to total (phosphorylated + unphosphorylated) Akt, ERK1/2 and p38, respectively. Cont, control (n = 4); Veh vehicle (n = 4–6); Resv resveratrol (n = 4–6). The data are presented as mean ± SEM. *p < 0.05 versus control; #p < 0.05 versus vehicle (ANOVA with posthoc Fisher’s PLSD test)
Resveratrol Increased PGC1α and pCREB Protein Levels
To determine whether the enhanced expression of SIRT1 and pAkt was associated with activation of their downstream molecules, we examined PGC1α and pCREB levels at 6 and 24 h after MCAO. PGC1α protein expression in the vehicle-treated cortex was unchanged at both time points relative to that seen in controls (Fig. 3a). However, PGC1α expression was markedly increased in the resveratrol-treated mice at 24 h (2.0-fold change relative to vehicle; p < 0.01). PGC1α mRNA level measured by real-time PCR (Fig. 3b) was also increased at 24 h after MCAO (1.9-fold change relative to vehicle; p < 0.01), indicating that resveratrol affects the expression of PGC1α in the ischemic cortex at both gene and protein levels.

Resveratrol effects on the levels of PGC1α protein (a) and mRNA (b), and pCREB (c) in the cortex at 6 and 24 h after MCAO. Values are expressed as ratios versus normal controls (given a nominal value of 1) at each time point after normalization. PGC1α was normalized to actin, and pCREB was normalized to total CREB. Cont, control (n = 4); Veh vehicle (n = 4–6); Resv resveratrol (n = 4–6). The data are presented as mean ± S.E.M. *p < 0.05 versus control; #p < 0.05 versus vehicle (ANOVA with posthoc Fisher’s PLSD test)
Similarly to those of PGC1α protein, pCREB levels were unchanged in the vehicle-treated cortex as compared to controls at both time points (Fig. 3c), but were significantly increased by resveratrol 24 h post-MCAO (1.29-fold change relative to vehicle; p < 0.01). These results indicate that increases of SIRT1 and pAkt levels by resveratrol lead to activation of their downstream molecules, PGC1α and pCREB.
Activation of Akt and CREB in Neurons by Resveratrol
We further examined whether Akt/CREB signaling was activated in neurons of the ischemic cortex at 24 h after MCAO using immunofluorescence staining. Expression of pAkt and pCREB was markedly increased in resveratrol-treated as compared to vehicle-treated brain tissue (Fig. 4a, b). Both of pAkt and pCREB were substantially co-localized with the neuron-specific marker NeuN. Also, localization of pCREB expressions in neuronal nuclei was verified by DAPI (Fig. 4b, right column). The results indicate that Akt/CREB signaling in neurons is activated by resveratrol.

Resveratrol increased expression of pAkt and pCREB in the ischemic cortex. Immunofluorescence staining was used to probe for pAkt (a; red), pCREB (b; red), and NeuN (green) 24 h after MCAO in the cortex of vehicle- or resveratrol-treated mice. Merged images are shown in the right column. Triple labeled (pCREB/NeuN/DAPI) cells were shown in an inserted box of the merged image in (b). Arrows indicated colocalizations of NeuN with pAKT or pCREB. An area of the ischemic cortex (red box) was used for image analysis as depicted in the pAkt and pCREB images. Veh vehicle, Resv resveratrol. Three sets of experiments were conducted independently. Representative images were obtained from one set. Scale bar = 50 μm in a and b; 20 μm in an inserted box of (b) (Color figure online)
Resveratrol Increased mRNA Levels of UCP2, SOD2, and Bcl-2
Finally, we examined expression changes in genes regulated by PGC1α and CREB 24 h after MCAO. Resveratrol significantly increased the expression of two mitochondrial anti-oxidative enzymes, UCP2 (1.51-fold change relative to vehicle; p < 0.05) and SOD2 (3.25-fold change relative to vehicle; p < 0.01), which are induced by PGC1α [17] (Fig. 5a–c). This finding is consistent with a previous report showing that PGC1α plays a role in neuroprotection by increasing the levels of both enzymes in the ischemic brain [31]. We also found that mRNA levels of the anti-apoptotic gene Bcl-2, a downstream target of CREB, were increased in the ischemic brain following resveratrol treatment (1.49-fold change relative to vehicle; p < 0.05; Fig. 5d). Altogether, these data suggest that resveratrol upregulates transcription of protective genes via modulation of the SIRT1/PGC1α and Akt/CREB pathways.

Resveratrol effects on mRNA levels of UCP2, SOD2 and Bcl-2 in the cortex 24 h after MCAO. a Reverse -transcribed PCR blots of UCP2, SOD2 and Bcl-2. Quantitative mRNA levels of UCP2 (b), SOD2 (c), and Bcl-2 (d) in control and ischemic cortices of vehicle- or resveratrol-treated mice (n’s = 3). Values are expressed as ratios of normal controls, which are given a nominal value of 1. Cont control, Veh vehicle, Resv resveratrol. The data are presented as mean ± SEM. *p < 0.05 versus control; #p < 0.05 versus vehicle (ANOVA with posthoc Fisher’s PLSD test)
Discussion
In this study, we demonstrate that neuroprotection afforded by resveratrol treatment during acute phase after MCAO is associated with modulation of multiple signaling pathways in the ischemic cortex. Resveratrol increased the levels of SIRT1/PGC1α/mitochondrial anti-oxidative enzymes, pAkt/pCREB/Bcl-2 and activation of p38, but inhibited the increase of pERK1/2.
Several studies have reported that resveratrol activates SIRT1 in various models of neurological disorders, but until now, the effects of resveratrol on SIRT1 have not been examined in the ischemic brain. The first evidence of neuroprotective effects of resveratrol being mediated via SIRT1 came from a study using a model of global ischemia [12]. Single pre-treatment with resveratrol 48 h before global ischemia increased SIRT1 activity transiently during the tolerance period and the increase was reversed by the SIRT1-specific inhibitor sirtinol [12]. However, SIRT1 activity was not tested during the ischemic period. The present study is the first to show increased SIRT1 protein levels in the ischemic cortex protected by resveratrol (Fig. 2a). A neuroprotective role for SIRT1 in vivo in the ischemic brain has recently been elucidated [18, 32]. SIRT1 protein levels are increased in the ischemic brain at 1 day after transient MCAO [18], as shown in our results (Fig. 2a), and SIRT1 levels are further enhanced by neuroprotective compounds, such as icariin and leptin when administered after MCAO [18, 32]. PGC1α is a target protein of SIRT1 and functions as a regulator of mitochondrial biogenesis and oxidative stress [17]. PGC1α induces many mitochondrial antioxidant enzymes in neurons exposed to hydrogen peroxide, and increasing PGC1α levels protect neuronal striatal cells from oxidative insult [17]. Because we previously showed that reactive oxygen species (ROS) production in the ischemic brain was reduced by resveratrol [9], it can be hypothesized that resveratrol induces ROS-detoxifying enzymes via SIRT1-dependent PGC1α expression. Consistent with this hypothesis, we found that resveratrol increased PGC1α mRNA and protein levels with induction of UCP2 and SOD2 in the ischemic cortex (Figs. 3a, b, 5a–c). Our results are in agreement with a recent in vivo study demonstrating mediation of resveratrol effects via SIRT1/PGC1α in a model of neurodegenerative disease [14]. The neuroprotective effect of resveratrol on dopaminergic neurons in a model of Parkinson’s disease was accompanied by increased expression of the antioxidant SOD2 in the substantia nigra [14]. Collectively, these findings suggest that increased expression of SIRT1/PGC1α with induction of ROS-detoxifying enzymes may underlie the anti-oxidative effect of resveratrol in the ischemic brain.
It has been reported that Akt plays a positive role in the protective effects of resveratrol in hippocampal slice cultures under oxygen-glucose deprivation (OGD) [33] and in neurons exposed to glutamate [34]. Resveratrol increases phosphorylation of Akt, which consequently leads to inactivation of glycogen synthase kinase 3β (GSK3β) activity, and resveratrol-mediated neuronal survival against OGD and glutamate is reversed by a phosphoinositide-3-kinase inhibitor [33, 34]. In the ischemic brain, there is considerable evidence that the Akt/CREB pathway is involved in neuronal survival [20, 35]. For example, neuroprotective reagents such as estrogen and cilostazol are associated with upregulation of pAkt/pCREB and anti-apoptotic Bcl-2 protein expression in the ischemic brain [21, 23], consistent with the findings of the present study (Fig. 2-5). It is generally accepted that apoptotic cell death rather than necrosis occurs in the ischemic penumbra, in which metabolic deficits are less severe because of collateral blood supply, as compared to the ischemic core [36]. Therefore, it is reasonable to infer that anti-apoptotic Bcl-2 upregulation through the Akt/CREB pathway contributes to the resveratrol-induced reduction in ischemic damage in the cortex, an area of penumbra (Fig. 1a). In light of our finding of increased gene levels of Bcl-2 (which localizes to the outer mitochondrial membrane) and mitochondrial anti-oxidative enzymes, mitochondria seem to be the main site of action of resveratrol against ischemic insult.
The literature on resveratrol modulation of ERK1/2 activity is inconsistent. For example, a study has reported that resveratrol increases pERK1/2 in hippocampal slice cultures exposed to OGD, although ERK1/2 inhibition fails to block resveratrol’s positive effects [33]. On the other hand, resveratrol prevented H2O2- and glutamate-induced ERK1/2 activation in hippocampal slices [26, 37], consistent with our finding that resveratrol blocked MCAO-induced increase in pERK1/2 (Fig. 2c). In ischemic stroke, increased ERK1/2 activity is believed to be involved in both neuroprotection and exacerbation of injury [24]. Sawe et al. speculated that pERK1/2 induced by stroke or other insults exaggerates ischemic injury by augmenting inflammation, whereas pERK1/2 induced by growth factors or neuroprotectants reduces ischemic injury by blocking apoptosis [24]. Because pERK1/2 was increased in the ischemic cortex at 24 h by ischemic stroke itself (Fig. 2c), the increase in pERK1/2 may have a detrimental effect on the ischemic brain in our model. Therefore, the inhibition of activated ERK1/2 may contribute to the anti-inflammatory effect of resveratrol such as the repression of IL-1β and TNF-α levels in the ischemic brain, as shown in our previous report [9]. In accordance with this view, a recent study showed that resveratrol downregulated ERK1/2 activation and thereby suppressed glutamate-induced expression of pro-inflammatory molecules, such as interleukin-1β and monocyte chemotactic protein-1in hippocampal slice cultures [26]. In contrast to pERK1/2, another MAPK p38 was increased by resveratrol (Fig. 2d). This was an unexpected result because of previous findings that activated p38 is associated with ischemic cell death and pp38 inhibition reduces ischemic injury [19]. In addition, it has been shown that resveratrol suppresses p38 activation in stimulated microglia and astrocytes [27–29, 38]. Although the mechanism by which activated p38 contributes to resveratrol-mediated neuroprotection is uncertain, recent studies suggest a positive role of activated p38 in the brain. Administration of a neuroprotectant icariin or sevoflurane preconditioning has been found to increase p38 phosphorylation in neurons protected from OGD or in the brain following ischemic stroke [39, 40]. The authors of this study suggest that icariin-induced p38 activation is involved in upregulation of SIRT expression [40]. Additionally, other studies have suggested that induction of brain-derived neurotrophic factor or pCREB by activated p38 in stimulated glia might contribute to neuronal survival or neuroplasticity [41–44]. Therefore, it is possible that activated p38 might have a role in the induction of SIRT1 and pCREB levels in resveratrol-mediated neuroprotection (Fig. 6).

Schematic diagram of the proposed mechanisms of resveratrol’s actions in the ischemic brain. In our model resveratrol enhances expressions of SIRT1/PGC1α, and activations of Akt/CREB and p38 MAPK. ERK1/2 activation is repressed in the ischemic brain. Dashed lines indicate possible interactions between signaling pathways (see text)
The results of previous studies suggest that there may be other types of cross-talk between signaling pathways in resveratrol-treated neurons or brains (Fig. 6). For example, resveratrol-associated PGC1α gene expression via SIRT1 relies on the transcription factor CREB in dopaminergic neurons [14], and CREB is involved in the regulation of the PGC1α gene promoter in neural cells under oxidative stress [17]. On the other hands, SIRT1 regulates CREB protein levels in neurons via a brain-specific microRNA, miR-134-mediated post-transcriptional mechanism [45]. In addition, PGC1α levels are stabilized by pAkt-mediated GSK3β inhibition [46], and pAkt promotes neuronal survival through suppression of ERK1/2 phosphorylation [47]. Therefore, it is likely that orchestrated modulation of signaling pathways contribute to resveratrol-induced neuroprotection. Because ischemic brain damage arises through diverse mechanisms, successful therapy may involve either a combination of drugs with different actions or the use of a single drug with multiple actions [48]. As a regulator of multiple pathways, resveratrol may be an ideal therapeutic agent for ischemic stroke. Because of resveratrol’s pleiotropic actions, it is as yet unknown which molecule is a key mediator of resveratrol-mediated neuroprotection against ischemic stroke. Although our findings suggest an association but not necessarily a causal relationship between signaling molecules and resveratrol-mediated neuroprotection, our study may serve as a foundation for future studies designed to identify a master regulator of resveratrol’s actions. Future mechanistic studies involving specific inhibitors of each pathway or transgenic mice will elucidate whether resveratrol directly or indirectly regulates the SIRT/PGC1α, Akt/CREB, and MAPK pathways in the ischemic brain.
In conclusion, the present study provides the first evidence that resveratrol upregulates the SIRT1/PGC1α, Akt/pCREB, and p38 pathways and downregulates pERK1/2 expression in a brain area protected by resveratrol from ischemic injury. These findings suggest that multiple signaling pathways contribute to resveratrol-mediated neuroprotection.
Abbreviations
- CREB:
-
Cyclic AMP-response-element-binding protein
- ERK1/2:
-
Extracellular signal-regulated kinase1/2
- GSK3β:
-
Glycogen synthase kinase 3β
- MAPK:
-
Mitogen-activated protein kinase
- MCAO:
-
Middle cerebral artery occlusion
- OGD:
-
Oxygen-glucose deprivation
- PGC1α:
-
Peroxisome proliferator-activated receptor γ coactivator 1α
- ROS:
-
Reactive oxygen species
- UCP2:
-
Uncoupling protein 2
- SOD2:
-
Superoxide dismutase 2
- SDS:
-
Sodium dodecyl sulfate
- SIRT1:
-
Sirtuin 1
- TBS:
-
Tris-buffered saline
References
Harikumar KB, Aggarwal BB (2008) Resveratrol: a multitargeted agent for age-associated chronic diseases. Cell Cycle 7:1020–1035
Baur JA, Sinclair DA (2006) Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov 5:493–506
Gao D, Zhang X, Jiang X, Peng Y, Huang W, Cheng G, Song L (2006) Resveratrol reduces the elevated level of MMP-9 induced by cerebral ischemia-reperfusion in mice. Life Sci 78:2564–2570
Huang SS, Tsai MC, Chih CL, Hung LM, Tsai SK (2001) Resveratrol reduction of infarct size in Long-Evans rats subjected to focal cerebral ischemia. Life Sci 69:1057–1065
Inoue H, Jiang XF, Katayama T, Osada S, Umesono K, Namura S (2003) Brain protection by resveratrol and fenofibrate against stroke requires peroxisome proliferator-activated receptor alpha in mice. Neurosci Lett 352:203–206
Lu KT, Chiou RY, Chen LG, Chen MH, Tseng WT, Hsieh HT, Yang YL (2006) Neuroprotective effects of resveratrol on cerebral ischemia-induced neuron loss mediated by free radical scavenging and cerebral blood flow elevation. J Agric Food Chem 54:3126–3131
Sinha K, Chaudhary G, Gupta YK (2002) Protective effect of resveratrol against oxidative stress in middle cerebral artery occlusion model of stroke in rats. Life Sci 71:655–665
Tsai SK, Hung LM, Fu YT, Cheng H, Nien MW, Liu HY, Zhang FB, Huang SS (2007) Resveratrol neuroprotective effects during focal cerebral ischemia injury via nitric oxide mechanism in rats. J Vasc Surg 46:346–353
Shin JA, Lee H, Lim YK, Koh Y, Choi JH, Park EM (2010) Therapeutic effects of resveratrol during acute periods following experimental ischemic stroke. J Neuroimmunol 227:93–100
Yousuf S, Atif F, Ahmad M, Hoda N, Ishrat T, Khan B, Islam F (2009) Resveratrol exerts its neuroprotective effect by modulating mitochondrial dysfunctions and associated cell death during cerebral ischemia. Brain Res 1250:242–253
Kelly GS (2010) A review of the sirtuin system, its clinical implications, and the potential role of dietary activators like resveratrol: part 2. Altern Med Rev 15:313–328
Della-Morte D, Dave KR, DeFazio RA, Bao YC, Raval AP, Perez-Pinzon MA (2009) Resveratrol pretreatment protects rat brain from cerebral ischemic damage via a sirtuin 1-uncoupling protein 2 pathway. Neuroscience 159:993–1002
Raval AP, Dave KR, Perez-Pinzon MA (2006) Resveratrol mimics ischemic preconditioning in the brain. J Cereb Blood Flow Metab 26:1141–1147
Mudo G, Makela J, Liberto VD, Tselykh TV, Olivieri M, Piepponen P, Eriksson O, Malkia A, Bonomo A, Kairisalo M, Aguirre JA, Korhonen L, Belluardo N, Lindholm D (2012) Transgenic expression and activation of PGC-1alpha protect dopaminergic neurons in the MPTP mouse model of Parkinson’s disease. Cell Mol Life Sci 69:1153–1165
Wang J, Zhang Y, Tang L, Zhang N, Fan D (2011) Protective effects of resveratrol through the up-regulation of SIRT1 expression in the mutant hSOD1-G93A-bearing motor neuron-like cell culture model of amyotrophic lateral sclerosis. Neurosci Lett 503:250–255
Albani D, Polito L, Batelli S, De Mauro S, Fracasso C, Martelli G, Colombo L, Manzoni C, Salmona M, Caccia S, Negro A, Forloni G (2009) The SIRT1 activator resveratrol protects SK-N-BE cells from oxidative stress and against toxicity caused by alpha-synuclein or amyloid-beta (1–42) peptide. J Neurochem 110:1445–1456
St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R, Spiegelman BM (2006) Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 127:397–408
Zhu HR, Wang ZY, Zhu XL, Wu XX, Li EG, Xu Y (2010) Icariin protects against brain injury by enhancing SIRT1-dependent PGC-1alpha expression in experimental stroke. Neuropharmacology 59:70–76
Irving EA, Bamford M (2002) Role of mitogen- and stress-activated kinases in ischemic injury. J Cereb Blood Flow Metab 22:631–647
Zhao H, Sapolsky RM, Steinberg GK (2006) Phosphoinositide-3-kinase/akt survival signal pathways are implicated in neuronal survival after stroke. Mol Neurobiol 34:249–270
Choi YC, Lee JH, Hong KW, Lee KS (2004) 17 Beta-estradiol prevents focal cerebral ischemic damages via activation of Akt and CREB in association with reduced PTEN phosphorylation in rats. Fundam Clin Pharmacol 18:547–557
Pugazhenthi S, Nesterova A, Sable C, Heidenreich KA, Boxer LM, Heasley LE, Reusch JE (2000) Akt/protein kinase B up-regulates Bcl-2 expression through cAMP-response element-binding protein. J Biol Chem 275:10761–10766
Lee JH, Kim KY, Lee YK, Park SY, Kim CD, Lee WS, Rhim BY, Hong KW (2004) Cilostazol prevents focal cerebral ischemic injury by enhancing casein kinase 2 phosphorylation and suppression of phosphatase and tensin homolog deleted from chromosome 10 phosphorylation in rats. J Pharmacol Exp Ther 308:896–903
Sawe N, Steinberg G, Zhao H (2008) Dual roles of the MAPK/ERK1/2 cell signaling pathway after stroke. J Neurosci Res 86:1659–1669
Kaminska B, Gozdz A, Zawadzka M, Ellert-Miklaszewska A, Lipko M (2009) MAPK signal transduction underlying brain inflammation and gliosis as therapeutic target. Anat Rec (Hoboken) 292:1902–1913
Lee EO, Park HJ, Kang JL, Kim HS, Chong YH (2010) Resveratrol reduces glutamate-mediated monocyte chemotactic protein-1 expression via inhibition of extracellular signal-regulated kinase 1/2 pathway in rat hippocampal slice cultures. J Neurochem 112:1477–1487
Zhong LM, Zong Y, Sun L, Guo JZ, Zhang W, He Y, Song R, Wang WM, Xiao CJ, Lu D (2012) Resveratrol inhibits inflammatory responses via the mammalian target of rapamycin signaling pathway in cultured LPS-stimulated microglial cells. PLoS ONE 7:e32195
Zhang F, Shi JS, Zhou H, Wilson B, Hong JS, Gao HM (2010) Resveratrol protects dopamine neurons against lipopolysaccharide-induced neurotoxicity through its anti-inflammatory actions. Mol Pharmacol 78:466–477
Bi XL, Yang JY, Dong YX, Wang JM, Cui YH, Ikeshima T, Zhao YQ, Wu CF (2005) Resveratrol inhibits nitric oxide and TNF-alpha production by lipopolysaccharide-activated microglia. Int Immunopharmacol 5:185–193
Lin TN, He YY, Wu G, Khan M, Hsu CY (1993) Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke 24:117–121
Chen SD, Lin TK, Yang DI, Lee SY, Shaw FZ, Liou CW, Chuang YC (2010) Protective effects of peroxisome proliferator-activated receptors gamma coactivator-1alpha against neuronal cell death in the hippocampal CA1 subfield after transient global ischemia. J Neurosci Res 88:605–613
Avraham Y, Davidi N, Porat M, Chernoguz D, Magen I, Vorobeiv L, Berry EM, Leker RR (2010) Leptin reduces infarct size in association with enhanced expression of CB2, TRPV1, SIRT-1 and leptin receptor. Curr Neurovasc Res 7:136–143
Zamin LL, Dillenburg-Pilla P, Argenta-Comiran R, Horn AP, Simao F, Nassif M, Gerhardt D, Frozza RL, Salbego C (2006) Protective effect of resveratrol against oxygen-glucose deprivation in organotypic hippocampal slice cultures: involvement of PI3-K pathway. Neurobiol Dis 24:170–182
Fukui M, Choi HJ, Zhu BT (2010) Mechanism for the protective effect of resveratrol against oxidative stress-induced neuronal death. Free Radic Biol Med 49:800–813
Du K, Montminy M (1998) CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem 273:32377–32379
Broughton BR, Reutens DC, Sobey CG (2009) Apoptotic mechanisms after cerebral ischemia. Stroke 40:e331–e339
de Almeida LM, Leite MC, Thomazi AP, Battu C, Nardin P, Tortorelli LS, Zanotto C, Posser T, Wofchuk ST, Leal RB, Goncalves CA, Gottfried C (2008) Resveratrol protects against oxidative injury induced by H2O2 in acute hippocampal slice preparations from Wistar rats. Arch Biochem Biophys 480:27–32
Zhou H, Chen Q, Kong DL, Guo J, Wang Q, Yu SY (2011) Effect of resveratrol on gliotransmitter levels and p38 activities in cultured astrocytes. Neurochem Res 36:17–26
Ye Z, Guo Q, Wang N, Xia P, Yuan Y, Wang E (2012) Delayed neuroprotection induced by sevoflurane via opening mitochondrial ATP-sensitive potassium channels and p38 MAPK phosphorylation. Neurol Sci 33:239–249
Wang L, Zhang L, Chen ZB, Wu JY, Zhang X, Xu Y (2009) Icariin enhances neuronal survival after oxygen and glucose deprivation by increasing SIRT1. Eur J Pharmacol 609:40–44
Cho J, Gruol DL (2008) The chemokine CCL2 activates p38 mitogen-activated protein kinase pathway in cultured rat hippocampal cells. J Neuroimmunol 199:94–103
Brautigam VM, Frasier C, Nikodemova M, Watters JJ (2005) Purinergic receptor modulation of BV-2 microglial cell activity: potential involvement of p38 MAP kinase and CREB. J Neuroimmunol 166:113–125
Rao JS, Ertley RN, Lee HJ, DeMar JC Jr, Arnold JT, Rapoport SI, Bazinet RP (2007) n-3 polyunsaturated fatty acid deprivation in rats decreases frontal cortex BDNF via a p38 MAPK-dependent mechanism. Mol Psychiatry 12:36–46
Yang H, Feng GD, Liang Z, Vitale A, Jiao XY, Ju G, You SW (2012) In vitro beneficial activation of microglial cells by mechanically-injured astrocytes enhances the synthesis and secretion of BDNF through p38MAPK. Neurochem Int 61(2):175–186
Gao J, Wang WY, Mao YW, Graff J, Guan JS, Pan L, Mak G, Kim D, Su SC, Tsai LH (2010) A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature 466:1105–1109
Clark J, Simon DK (2009) Transcribe to survive: transcriptional control of antioxidant defense programs for neuroprotection in Parkinson’s disease. Antioxid Redox Signal 11:509–528
Subramaniam S, Shahani N, Strelau J, Laliberte C, Brandt R, Kaplan D, Unsicker K (2005) Insulin-like growth factor 1 inhibits extracellular signal-regulated kinase to promote neuronal survival via the phosphatidylinositol 3-kinase/protein kinase A/c-Raf pathway. J Neurosci 25:2838–2852
Fisher M (2011) New approaches to neuroprotective drug development. Stroke 42:S24–S27
Acknowledgments
This study was supported by a grant of the Korea Healthcare technology R&D Project, Ministry for Health, Welfare & Family Affairs, Republic of Korea (A080078) and by Mid-career Researcher program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (MEST) (2011-0015923) to E.M.P., and by NRF grant funded by the MEST (2010-0029354) to H.S.K.
Conflict of interest
The authors report no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shin, J.A., Lee, KE., Kim, HS. et al. Acute Resveratrol Treatment Modulates Multiple Signaling Pathways in the Ischemic Brain. Neurochem Res 37, 2686–2696 (2012). https://doi.org/10.1007/s11064-012-0858-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-012-0858-2