
Abstract
Diabetes mellitus, a debilitating chronic disease, affects ~100 million people. Peripheral neuropathy is one of the most common early complications of diabetes in ~66 % of these patients. Altered Ca2+ handling and Ca2+ signaling were detected in a huge variety of preparations isolated from animals with experimentally induced type 1 and 2 diabetes as well as patients suffering from the disease. We reviewed the role of Ca2+ signaling through cation channels and oxidative stress on diabetic neuropathic pain in sensory neurons. The pathogenesis of diabetic neuropathy involves the polyol pathway, advanced glycation end products, oxidative stress, protein kinase C activation, neurotrophism, and hypoxia. Experimental studies with respect to oxidative stress and Ca2+ signaling, inhibitor roles of antioxidants in diabetic neuropathic pain are also summarized in the review. We hypothesize that deficits in insulin, triggers alterations of sensory neurone phenotype that are critical for the development of abnormal Ca2+ homeostasis and oxidative stress and associated mitochondrial dysfunction. The transient receptor potential channels are a large family of proteins with six main subfamilies. The sheer number of different TRPs with distinct functions supports the statement that these channels are involved in a wide range of processes ranging in diabetic neuropathic pain and it seems that the TRPC, TRPM and TRPV groups are mostly responsible from diabetic neuropathic pain. In conclusion, the accumulating evidence implicating Ca2+ dysregulation and over production of oxidative stress products in diabetic neuropathic pains, along with recent advances in understanding of genetic variations in cation channels such as TRP channels, makes modulation of neuronal Ca2+ handling an increasingly viable approach for therapeutic interventions against the painful and degenerative aspects of many diabetic neuropathies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diabetic neuropathy in Type 1 and 2 diabetes in both humans and animals models is associated with reduction of motor and sensory nerve conduction velocity and structural changes in peripheral nerve including endoneural microangiopathy, abnormal Schwann cell pathology, axonal degeneration, paranodal demyelination and loss of myelinated and unmyelinated fibers- the alter [1].
Peripheral neuropathy is one of the most serious complications of diabetes. Peripheral neuropathy is one of the most common early complications of diabetes in ~66 % of these patients. Diabetic neuropathy is frequently painful that typically involves the extremities occurring as an exaggerated response to either a painful stimulus (hyperalgesia) or a mild and normally non-painful stimulus (allodynia) [2, 3]. Existing therapies for this devastating complication of diabetes are largely inadequate.
Neuropathic pain manifesting itself as allodynia and hyperalgesia continues to be a significant problem in clinical medicine. Increasing the local Ca2+ concentration at the site of injury or in the spinal cord may contribute to the development or neuropathic pain [4]. The main routes of extracellular Ca2+ influx to the cells are voltage-dependent Ca2+ channels. Current arising from voltage-dependent Ca2+ channels are subdivided into 2 major classes based on the membrane potential at which they become activated: high-voltage activated or sustained currents which are further divided into I, P, O, N and R subtypes and low-voltage activated or transient (T-type) Ca2+ currents which are further divided into Cav 3.1, Cav 3.2, Cav 3.3 [5].
When pain messages are transmitted from the periphery to the central nervous system, the nociceptive transmitters such as substance P are released via exocytosis from the primary sensory terminals in the spinal dorsal horn, which is regulated by high threshold voltage-dependent Ca2+ channels such as N-and P/Q-types located in presynaptic nerve terminals [5]. So far, several studies including our previous report have demonstrated that voltage-dependent Ca2+ channels may contribute to the streptozotocin (STZ)-induced diabetic neuropathic pain [6]. The increased responsiveness of the spinal pain transmission is likely due to the increased responsiveness of the primary afferent neurons, which could results in enhanced neurotransmitter exocytosis via the opening of voltage-dependent Ca2+ channels or due to the postsynaptic hyperexcitability in dorsal horn projection neurons, which possible to be induced by enhanced Ca2+-influx through voltage-dependent Ca2+ channels. There are 6 subfamilies of transient receptor potential (TRP) channels and oxidative stress-dependent Ca2+ over influxes through the TRP channels have also important role in diabetes and diabetic neuropathic pain. Such changes in transmission within the spinal cord may contribute to diabetic neuropathic pain [6].
The human body is equipped with a complete arsenal of defenses against external and internal aggressions. Those against the so-called reactive oxygen species (ROS) such as superoxide anion, hydroxyl radical and hydrogen peroxide are crucial in inflammatory responses where they participate in physiological processes such as the arachidonic acid cascade and phagocytosis [7]. It is generally believed that oxidative stress is the key pathological process inducing nerve damage in diabetes [8]. The following pathways, triggered by hyperglycaemia and/or lack of insulin, contribute to oxidative stress in diabetic neuropathy:
-
1.
poly pathway-excess glucose drives increased aldose reductase activity resulting in elevated flux through the poly pathway and build up damaging levels of polyols and associated pro-oxidants [9]. High intracellular glucose concentration also directly elevates ROS through raised Ca2+ influx and mitochondrial activity [10];
-
2.
protein glycation-hyperglycaemia induced these proteins and/or activation of the receptor for advanced glycation end-products (RAGE) which induces cellular stress, in part, through activation of inflammatory pathways [9], and
-
3.
reduced neurotrophic support-maintenance of normal phenotype of sensory neurons is impaired due to diabetes-induced loss of neurotrophic support by insulin and the insulin-like growth factors (IGF-1, IGF-2), nerve growth factor (NGF) and neurotrophin-3 (NT-3) [11].
Mitochondria are main intracellular source of ATP production. They also involved many other cellular functions including Ca2+ signaling and apoptosis. Mitochondria are also major sites for reactive oxygen species (ROS) and reactive nitrogen species productions [5]. In β-cells it has been reported that ROS, and probably hydrogen peroxide in particular, are one of the metabolic coupling factors in diabetes [12]. Moreover exposure of β-cells to low glucose activates 5′ AMP-activated protein kinase (AMPK) in a superoxide-dependent, AMP-independent way [13]. Hence, ROS may contribute to physiological and pathophysiological control of β-cell functions.
In the review papers, we reviewed last development of mitochondria, endoplasmic reticulum and TRP channels dependent-Ca2+ signaling and oxidative damage in neuropathic pain.
Diabetic Neuropathic Pain and Ca2+ Signaling
Diabetes mellitus, a debilitating chronic disease, affects ~100 million people [8]. Therapies for this devastating complication of diabetes are largely inadequate, partly attributable to lack of insight into the pathophysiological mechanisms of this disease [9]. One of the most prominent features of diabetic peripheral neuropathy is the development of pain that typical involves the extremities, occurring as an exaggerated response to either a painful stimulus (hyperalgesia) or a mild and normally nonpainful stimulus (allodynia). The precise cellular mechanisms of hyperalgesia and allodynia neuropathic pain remain poorly understood, but the remodeling of voltage-and ligand-gated ion channels that can increase excitability of the sensory neurons may play a critical role [9, 10].
Glucose entry into β-cell promotes glycolysis, generating pyruvate that is imported into mitochondria, where it feeds the tricarboxylic acid (TCA) cycle. TCA cycle activation induced transfer of electrons from TCA cycle intermediates to the respiratory chain reactions via NADH and FADH2 and then the chain reactions induce production of ROS in mitochondria. The mitochondrial electron transport chain is an important site of ROS production within the cell. Electrons from sugar, fatty acids, and amino acids catabolism accumulate in the electron carriers of the respiratory chain reactions. ROS formation is coupled to electron transport as byproducts of normal mitochondrial respiration through the one-electron reduction of molecular oxygen [14]. In addition, mitochondria are not the only contributes ROS generation in pancreatic islets. Indeed, β-cells express phagocyte like NADPH oxidases, negatively modulating the secretary response by reducing cAMP secondary to ROS generation [15]. Moreover, NADPH oxidases have been reported to be responsible from Rac1-Nox-ROS-JNK1/2 signaling pathway in the islet β-cell leading to the onset of mitochondrial dysregulation in the T2D zucker diabetic fatty rats.
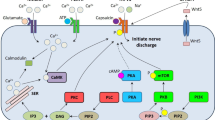
In diabetes, alterations in neuronal Ca2+ signaling may contribute to the development of distal symmetrical sensorimotor polyneuropathy and pain [11] (Table 1). Diabetes increases the current amplitude of multiple voltage-dependent Ca2+ currents [1], and Ca2+ influx activates nitric oxide (NO) cGMP/protein kinase G pathways; blockade of this pathways decreases experimentally induced pain [40]. In diabetic rats, protein kinase A (PKA), protein kinase C (PKC), and NO second messenger systems contribute to hyperalgesia, whereas N-methyl-d-asparate (NMDA) receptor-mediated events are not thought to be involved [16] (Fig. 1). Additionally, persistent elevations of cytosolic Ca2+ have been implicated in neuronal degeneration [17] and apoptosis [18]. Therefore, attenuation of oxidative stress and amelioration of abnormal Ca2+ signaling have emerged as important therapeutic targets in sensorimotor polyneuropathy [19].

Cells regulate intracellular Ca2+ levels lightly and excessive Ca2+ loads can lead to inappropriate activation of process that are normally operate at low levels, causing metabolic derangements and eventual cell death. Excessive Ca2+ load, in particular via N-methyl-d-aspartate (NMDA) receptors (NMDAR), is toxic to neurons in neurodegenerative diseases. NMDAR-mediated Ca2+ entry triggers a neurotoxic signal cascade involving the activation of neuronal nitric oxide (NO) synthase (nNOS), formation of the toxic ROS and NO and activation of the pro-apoptotic proteins poly(ADP-ribose) polymerase (PARP-1). Diabetes and hyperglycelima stimulate Ca2+ influx into cytosol through TRP channels by activation of ROS production. Sustained depolarization of mitochondrial membranes and enhanced ROS production activates transient receptor potential (TRP) channels such as TRP melastatin 2 (TRPM2), TRP vanilloid (TRPV) and voltage gated Ca2+ channels (VGCC) and Ca2+ influx increases by activation of TRP via ROS. The molecular pathway may be a cause of neurological symptoms and represents a fruitful subject for further study
A great body of evidence has been gathered about diabetes-induced changes of calcium signaling in different cell types. It has been reported that current densities of L- type Ca2+ channels were increased in diabetic beta cells [4]. Diabetes mellitus can affect both the peripheral and central nervous system. It has been shown that depolarization-induced Ca2+ transients in small, but not in large, dorsal root ganglion (DRG) neurons became substantially prolonged during STZ-induced and spontaneously occurring diabetes [5]. High-threshold, voltage-dependent calcium currents were enhanced in acutely dissociated, capsaicin-sensitive DRG neurons from diabetic rats, compared with non-diabetic controls [20]. Diabetes-induced alterations in neuronal Ca2+ homeostasis might explain the observed differential effects of diabetes on long-term potentiation and long-term depression in rat hippocampus.
Umeda et al. [6] investigated the gene and protein expression levels of α1 subunits in the dorsal root ganglia and the spinal cord from STZ-induced diabetic mice using real-time polymerase chain reaction and immunohistochemistry. They found that the STZ-induced diabetic hyperalgesia may be caused by a selection alteration in expression of P/Q-type voltage-dependent Ca2+ channels in mouse DRG neurons especially in small C-fibers and medium. They concluded that an enhanced expression of P\Q-type channels results in an increased transmitter release at the primary afferent neurons, which may lead to nociceptive abnormality.
Mibefradil, a benzimidazolyl-substituted tetraline derivative, is a novel Ca2+ channel blocker with several interesting pharmacological properties. Previous research has indicated that mibefradil may act on peripheral T-type Ca2+ channels in antinociceptive effects in neuropathic pain. Recently, Wen et al. [21] reported that there were T-type Ca2+ channels (Cav3.2 and Cav3.3, not Cav3.1) in the lumbar spinal cord of rats, and under the neuropathic pain conditions, the mRNA expression of T-type Ca2+ channels increased. They concluded that T-type Ca2+ channels may play an important role to the development of the neuropathic pain following chronic compression of DRG neuron operation.
Li and Chen [22] PKCβ is a serine/threonine kinase that is activated by intracellular Ca2+ and may, in turn, modify Ca2+ signaling. The Ca2+ homeostasis in excitable cells such as neurons is tightly regulated because intracellular Ca2+ is a pivotal second in many signaling cascades and is linked to such physiological phenomena as neurotransmitter release, cell survival, and axonal growth and maintenance. Its disturbance is a critical issue in neurodegenerative diseases. Similarly, the abnormality of Ca2+ homeostasis in diabetic DRG neurons has been shown to be an early molecular marker of diabetic neuropathy. The enhanced voltage-dependent Ca2+ current and impaired inhibitory G-protein function, and derangement of intracellular organelles with a Ca2+ buffering effect, such as endoplasmic reticulum and mitochondria have been shown to contribute to disturbed calcium signaling in diabetic neuropathy.
The pathogenesis of diabetic neuropathy involves the polyol pathway, advanced glycation end products, oxidative stress, PKC activation, neurotrophism, and hypoxia. These factors may contribute to the development of nerve dysfunction and degeneration [4–7, 10]. Among them, the activity of PKC, especially its β isoform, may play a major role. On the basis of its pathogenic mechanism, PKCβ selective inhibitor LY333531 is currently under investigation in clinical trials. Tahara et al. [23] investigated the possible direct action of LY333531 on DRG neurons to intracellular Ca2+ homeostasis. They demonstrated that the PKCβ selective inhibitor LY ameliorates disturbed Ca2+ homeostasis via decreased mitochondrial Ca2+ buffering in small DRG neurons of diabetic rats.
Kostyuk et al. [24] in 1999 published work showing elevated resting [Ca2+]i in small neurons of the DRG of type 1 and type 2 diabetic mice, however, observed no changes in large neurons and in a follow-up study found no change in cytosolic free Ca2+ ([Ca2+]i) concentrations in any DRG cell type of STZ-diabetic rodents. Specifically, Kostyuk et al. [24] also demonstrated in small DRG neurons isolated from STZ-treated C57Blac6 mice, a model of type 1 diabetes, the resting [Ca2+]i was ~30 % higher than in control (250 ± 16 nm vs. 156 ± nm); the elevation in [Ca2+]i in small neurons was even greater in type 2 diabetic db/db mice. In their study, there were no differences in [Ca2+]i concentrations in large neurons, however, in either mouse model. It has to be noted though, that the same group, almost at the same time, reported the absence of any changes in resting [Ca2+]i in DRG neurons isolated from type 1 STZ-diabetic rodents; the reason for such a discrepancy remain unknown but may relate to differences in severity and/or length of STZ-diabetes within these studies.
In study of Drel et al. [25] it appears that the db/db mice the (type 2 diabetes) may have been maintained for 2–3 months. In STZ-diabetic Wistar rats of 8–14 weeks duration resting [Ca2+]i concentrations was substantially increased by 2–2.5 fold in both large and small DRG neurons isolated from lumbar L4–L5 DRG; the [Ca2+]i concentrations increase correlated with the progression of the disease. Conversely, resting [Ca2+]i was not affected by 8–14 weeks of experimental diabetes in neurons from ganglia located at higher levels of the spinal cord (C3–C4).
Jagodic et al. [9] found that T-type channels in medium-size DRG neurons in STZ-induced diabetic neuropathy show prominent changes in voltage-dependent in activation, allowing a greater fraction of the channels to be available for activation during both short and prolonged periods of depolarization.
Voitenko et al. [1] investigated effects of STZ-induced diabetes on neuronal. They measured the [Ca2+]i concentrations via Fura-2AM Ca2+ signaling method in dorsal horn neurons from acutely isolated spinal slices. They found that the K+-induced [Ca2+]i elevation was inhibited to a different extent by nickel ions, nifedipine and ω-conotoxin suggesting the co-expression of different subtypes of plasmalemmal voltage-gated Ca2+ channels. In their study, the suppression of [Ca2+]i transients by Ni2+ (50 μM) was the same in control and diabetic neurons. On the other hand, inhibition of [Ca2+]i transients by nifedipine (50 μM) and ω-conotoxin (1 μM) was much greater in diabetic neurons compared with normal animals.
The endoplasmic reticulum plays an important role in multiple programmed cell death pathways. Apoptosis caused by endoplasmic reticulum-induced oxidative stress has been also related with diabetes [26, 27] and can be caused by the accumulation of unfolded proteins resulting from disrupted Ca2+-dependent changes in the endoplasmic reticulum [28]. Both thapsigargin, a potent and specific inhibitor of sarco-endoplasmic reticulum ATPase (SERCA), and endogenous factors that downregulate SERCA, evoke endoplasmic reticulum-induced oxidative stress and apoptosis in β-cells [29, 30].
In addition to multiple isoforms of SERCA, the endoplasmic reticulum of β-cells express several classes of cytosolic Ca2+-releasing channels including the inositol triphosphatase receptors (IP3Rs) and ryanodine receptors [31, 32]. In the oxidative stress-induced diabetic state, the expressions of these receptors are known to be modulated in several cell types including pancreatic β-cells [33]. It was recently reported that ryanodine receptors inhibition reduced the ratio of ATP to ADP in MIN6 β-cells, an event that could conceivability activate endoplasmic reticulum oxidative stress [34]. Furthermore, studies of other cells have indicated that endoplasmic reticulum oxidative stress-related damage can be affected by inhibitors of IP3Rs and ryanodine receptors [35].
Neuropathic Pain, Ca2+ Signaling, Oxidative Stress and Mitochondria
The human body is equipped with a complete arsenal of defenses against external and internal aggressions. Those against the so-called reactive oxygen species (ROS) such as superoxide anion, hydroxyl radical and hydrogen peroxide are crucial in inflammatory responses where they participate in physiological processes such as the arachidonic acid cascade and phagocytosis [7]. Mitochondria are the major sources of ROS production since unpaired electrons are generated in the process of oxidative phosphorylation. Partial reduction of molecular oxygen by the unpaired electrons leads to the production of superoxide radicals which are one of the ROS and readily converted to hydrogen peroxide by superoxide dismutase enzymes [36]. There are three types of superoxide dismutase enzymes and magnesium superoxide dismutase enzyme is present in mitochondria matrix [28]. Unlike hydrogen peroxide, superoxide anion does not pass that readily across the membrane and thus for superoxide radicals produced in the matrix, the activity of manganese dependent superoxide dismutase enzyme is critical to prevent the mitochondrial matrix components for oxidative stress.
Tricarboxylic acid (TCA) cycle and electron transport chain contribute to key enzymatic components of mitochondria. During the process of braking down carbon substrates into acetyl CoA, reducing equivalents are produced, which are then fed to the electron transport chain consisting Complex I (NADH dehydrogenase), Complex II (succinate dehydrogenase), Complex III (ubiquinol cytochrome c reductase), and Complex IV (cytochrome c oxidase). Complex V is ATP synthase or proton-translocating ATP synthase [37]. Complexes I-IV involve ubiquinone (Coenzyme Q10). On the other world, they contain flavins, which contain riboflavin, iron-sulfur clusters, copper centers, or iron-containing heme moieties. Complex I is the main site for mitochondrial ROS production, where superoxide radicals are produced on the matrix side and rapidly dismutated to hydrogen peroxide [25, 39]. In addition, Complex III has also been reported as a site of superoxide radical production [25]. Complex III has important role on ischemic and apoptotic superoxide radical production.
The concentrations of ROS are kept under strict control by the activity of a complex defense system including enzymes and non-enzymatic species such as vitamin C, vitamin E, vitamin A or β-carotene [8]. Vitamin E, α-tocopherol, is the most important antioxidant in the lipid phase of cells. Vitamin E acts to protect cells against the effects of free radicals, which are potentially damaging byproducts of the body’s metabolism [38]. Vitamin C, as well as being a free radical scavenger, also transforms vitamin E to its active form. These enzymatic and non enzymatic antioxidants are also essential for inhibition of phagocytic activity related to ROS production [7, 8, 38]. Hydrogen peroxide is converted to water by glutathione peroxidase and it is one of the body’s major antioxidants [7].
Complex I is especially susceptible to NO damage, and animals administrated natural and synthetic complex I antagonists have undergone death of neurons [39, 40]. Increased oxidative and nitrosative stresses [41] have also emerged as leading candidates in the pathogenesis of distal symetrical sensorimotor polyneuropathy. A direct relationship has emerged between measures of oxidative stress and the development of nerve blood flow and nerve conduction deficits [42–45] as well as impaired neurotrophism [46, 47]. Oxidative stress has also recently been invoked as a contributing factor to painful symmetrical sensorimotor polyneuropathy [48]. However attenuation of oxidative stress alone may be insufficient to completely alleviate neuropathic pain complicating diabetes, emphasizing the need to impact additional metabolic pathways implicated in pain pathogenesis [49].
Taurine (2-aminoethanesulfonic acid) functions as in important endogenous antioxidant [50] and it is well known that Ca2+ modulator and neurotransmitter could promote chronic cytotoxicity in diabetes. They aimed to explore this hypothesis by assessing the potential of taurine replacement to ameliorate hyperalgesia and abnormal Ca2+ signaling in sensory neurons of STZ-induced diabetic rats and they found that taurine replacement may provide a novel mechanistically based approach to the treatment or prevention of distal symmetrical sensorimotor polyneuropathy [18].
Role of Transient Receptor Potential (TRP) Channels in Diabetes
The trp gene was identified through genetic studies of a mutation in the Drosophilia visual transduction system. The term of “TRP” is derived from “transient receptor potential”, because photoreceptors with the trp gene mutant fail to generate the Ca2+-dependent “sustained” phase of receptor potential and therefore fail to show subsequent Ca2+-dependent adaptation to light. There are 30 mammalian TRP channels, grouped into six subfamilies. Members of the TRP channels superfamily include TRP canonical (TRPC) subfamily consisting of 7, TRP vanilloid (TRPV) subfamily consisting of 6, TRP melastatin (TRPM) subfamily consisting of 8, TRP polycystein (TRPP) subfamily consisting of 3, TRP mucolipin (ML) subfamily consisting of 3, and TRP ankyrin (TRPA) subfamily consisting of only one member [51, 52].
TRPM2 Channels
TRPM2 may be a new target for diabetes therapy (Table 2). In study of Uchida and Tominaga [53], they observed impaired glucose tolerance and impaired insulin secretion in TRPM2 knockout mice. In addition, insulin secretion via TRPM2 occurs through control of intracellular Ca2+ concentrations and Ca2+ influx-independent mechanisms. It was recently observed that basal blood glucose levels increased in TRPM2-KO mice as compared to wild-type mice without any difference in plasma insulin levels. In isolated β-cells, smaller intracellular Ca²+ increase was observed in response to high concentrations of glucose in TRPM2-KO cells than in wild-type cells. Moreover, insulin secretion from the islets of TRPM2-KO mice in response to glucose treatment was impaired, whereas the response to tolbutamide, an ATP-sensitive potassium channel inhibitor, was not different between the two groups. They concluded that TRPM2 is involved in insulin secretion stimulated by glucose [54]. Romero et al. [55] investigated gene variation of TRPM2 in the pathophysiology of type 2 diabetes mellitus and they observed no evidence for an association of the variants tested with T2DM, although HOMA-%B was negatively associated with three TRPM2 variants (rs2838553, rs2838554 and rs4818917). Qian et al. [56] found that a current in βTC3 insulinoma cells and provided a mechanism for oscillatory calcium responses in the presence of glucose.
TRPV1 Channels
In study of Wei et al. [57] reported that in the sensory nerve fibers, could modulate the cardiac function, be impaired by diabetes and could contribute the further severe postischemic heart injury. Bishnoi et al. [58] found that STZ induced a direct effect on neurons trough expression and function of the TRP vanilloid 1 (TRPV1) channel in sensory neurons resulting in thermal hyperalgesia, even in non-diabetic STZ-treated mice. In the present study, they investigated the role of expression and function of TRPV1 in the central sensory nerve terminals in the spinal cord in STZ-induced hyperalgesia in rats. It was reported that both STZ-and transgene-mediated T1D are associated with two distinct phases of thermal pain sensitivity that parallel changes in TRPV1 as determined by paw withdrawal latency [59]. To understand the mechanism underlying this phenomenon, DRG neurons and stably TRPV1 expressing human embryonic kidney (HEK)293 cells, Pabbidi et al. [59] investigated the expression and function of TRPV1. They found that STZ has a direct action on neurons and modulates the expression and function of TRPV1, a nociceptive ion channel that is responsible for inflammatory thermal pain. Zsombok et al. [60] tested the hypothesis that synaptic modulation by TRPV1 receptors is reduced in the DMV in slices from a murine model of type 1 diabetes. The TRPV1 agonist capsaicin robustly enhanced glutamaterelease onto DMV neurons by acting at pre-terminal receptors in slices from intact mice, but failed to do so in slices from diabetic mice [60]. A recent study of Wilder-Smith et al. [61] suggested that in human painful neuropathies, epidermal TRPV1 expression is mainly in keratinocytes. Ohanyan et al. [62] subjected mice lacking TRPV1 [TRPV1(-/-)], db/db, and control C57BLKS/J mice to in vivo infusion of the TRPV1 agonist capsaicin or the α-adrenergic agonist phenylephrine to examine the integrated circulatory actions of TRPV1. TRPV1(-/-) mice exhibited no changes in MAP in response to capsaicin, suggesting the actions of this agonist are specific to TRPV1 activation [62]. Manni et al. [63] their results point to the potential of electro-acupuncture as a supportive therapy for the treatment of diabetic neuropathies. The efficacy of electro-acupuncture might depend on its actions on spinal/peripheral NGF synthesis/utilization and normalization of the levels of several sensory neuromodulators. Hyperglycemia and hypoxia are two main phenomena in diabetes associated with several complications [63] and Ristoiu et al. [64] found that hypoxia is a new sensitization mechanism for TRPV1, which might be relevant to diabetes-related complications, and also for other diseases that are associated with acute hypoxia.
Tanaka et al. [65] their study is the first to show the anti-diabetic pharmacological effects of the TRPV1 signal inhibitor N-(4-tertiarybutylphenyl)-4-(3-cholorphyridin-2-yl)tetrahydropyrazine-1(2H)- carbox-amide (BCTC). These findings suggest that TRPV1 antagonists may represent a new class of drugs effective in treating type 2 diabetes mellitus because of their dual effects as insulin sensitizers and secretagogues [65]. Liu et al. [66] examined whether cardiac nociception was altered in the STZ-induced diabetic rat model by assessing intrapericardial capsaicin-evoked electromyography responses in the spinotrapezius muscle. Their results suggested that STZ-induced diabetic rats develop somatic mechanical allodynia, but reduced cardiac nociception. They concluded that decreased TRPV1 function may contribute to the reduction of cardiac nociception in the diabetic rat [66]. The up regulation of kinin B1R in spinal dorsal horn microglia by pro-inflammatory cytokines is proposed as a crucial mechanism in early pain neuropathy in STZ-diabetic rats [67]. Mohammadi-Farani et al. [68] aimed to see if diabetic hyperalgesia is related to changes in TRPV1 or Cannabinoid CB1 receptors of periaqueductal gray. Kang et al. [69] investigated whether dietary capsaicin can reduce obesity-induced inflammation and metabolic disorders such as insulin resistance and hepatic steatosis. Facer et al. [70] reported that human DRG sensory neurons co-expressed TRPV1 and TRPV3, and that these were increased in injured human DRG. Related receptors TRPV4, activated by warmth and eicosanoids, and TRPM8, activated by cool and menthol, have been characterized in pre-clinical models [52, 70]. However, the role of TRPs in common clinical sensory neuropathies needs to be established.
TRPM6 and TRPM7 Channels
Romero et al. [55] hypothesized that gene variation of TRPM6 and TRPM7 may play a role in type 2 diabetes mellitus. Using a case–control population sample of the Boston metropolitan area (all whites, 455 controls and 467 cases), they assessed the relationship of 29 TRPM6 and 11 TRPM7 tag-single nucleotide polymorphisms (SNPs) with (1) several diabetes-related intermediate phenotypes (fasting insulin levels, fasting glucose levels, hemoglobin A1c, and homeostatic model assessment) and (2) the presence of T2DM. Wuensch et al. [71] investigated the effects of high glucose-induced oxidative stress on TRP channel expression in human monocytes and they were observed that Increased TRPC3 and TRPC6 protein expression was accompanied by increased 1-oleoyl-2-acetyl-sn-glycerol-induced calcium influx, which was blocked by the TRPC inhibitor 2-aminoethoxydiphenylborane (2-APB). Romero et al. [72] reported that TRPM7 gene variation might play a role in the risk of ischemic stroke. Results of Song et al. [73] provided suggestive evidence that two common non-synonymous TRPM6 coding region variants, Ile1393Val and Lys1584Glu polymorphisms, might confer susceptibility to type 2 diabetic women with low magnesium intake. Landman et al. [74] found that the TRPM7-associated Mg2+-inhibited cation (MIC) channel underlies ion channel dysfunction in presenilin FAD mutant cells, and the observed channel deficits are restored by the addition of PIP2, a known regulator of the MIC/TRPM7 channel.
TRPC Channels
Mita et al. [75] reported that Ca2+ entry from the extracellular space via alpha (1)—adrenoceptor-activated, Ca2+-permeable channels, but not voltage-gated Ca2+ channels, is impaired in endothelium–denuded caudal artery smooth muscle from type 2 diabetic Goto-kakizaki rats. Hence, TRPC channel expression may be responsible in part, for the dysfunction of receptor-mediated Ca2+ entry in caudal artery smooth muscle of Goto-Kakizaki rats. Recently a study was performed to investigate the underlying mechanism, particularly the roles of ROS and protein kinase C, in the diabetes induced TRPC6 down regulation [76] and it was found that high glucose significantly reduced TRPC6 protein expression in cultured mesangial cells [76]. Zbidi et al. [77] reported that expression of TRPC3, Orail and STIM1 is enhanced in DM2 subjects as compared to controls. Their findings provide an explanation to the enhanced Ca2+ entry induced by physiological agonists in platelets from diabetes mellitus patients with type 2 [77]. Chung et al. [78] reported that diabetes would modulate the capacitative calcium entry likely through the store-operated calcium channel specifically via the regulation of TRPC. In a study of Liu et al. [79], high glucose increases TRPC6 channel protein expression on the platelet surface which is mediated by a phosphatidylinositol 3-kinase-dependent pathway. Niehof and Borlak [61] reported dysregulation of HNF4 alpha and TRPC1 as a possible molecular rationale in diabetic nephropathy.
Conclusions
Diabetes induces a distal symmetrical polyneuropathy in which sensory dysfunction is an early and frequent feature. A number of pathogenic pathways secondary to hyperglycaemia, namely polyol pathway flux, oxidative stress, impaired neurotrophic factos and advanced glycation end-products have been proposed for the molecular basis of diabetic neuropathy and pain. Based on the results discussed above, we suggest that hyperglycaemia-dependent alterations of Ca2+ influx through cation channels, mitochondrial function and oxidative stress induced parallel pathophysiological mechanism in diabetic neuropathy. In this mechanism, impaired insulin signaling and Ca2+ influx through TRP and voltage gated Ca2+ channel activations triggers sensory neuron mitochondrial depolarization. The consequent increase in mitochondrial depolarization induces further ROS production and disrupts Ca2+ homeostatic mechanisms, particularly voltage gated Ca2+ channels.
Mutations in TRPs are linked to pathophysiology of diabetic neuropathy. Progression from mutations in TRPs to pathophysiology and diabetes will cause understanding of etiology of neuropathic neuropathy. In this review, we also focused on two distinct aspects of TRP channel physiology, the role of TRP channels in intracellular Ca2+ homeostasis, and their role in the transduction of diabetic pain in sensory neurons and it seems that the TRPC, TRPV and TRPM groups are mostly responsible from diabetic neuropathic pain. There are scarce reports some TRP channels such as TRPM2 channels in diabetic neuropathic pain. In future, role of other TRP channels such as TRPM2 on diabetic DRG neurons should have investigated in animal and human models.
Abbreviations
- 2-APB:
-
Aminoethoxydiphenylborane
- HEK:
-
Human embryonic kidney
- IGF:
-
Insulin growth factor
- NGF:
-
Nerve growth factor
- NMDA:
-
N-methyl-d-asparate
- NO:
-
Nitric oxide
- PKA:
-
Protein kinase A
- PKC:
-
Protein kinase C
- RAGE:
-
Advanced glycation end-products
- ROS:
-
Reactive oxygen species
- SERCA:
-
Sarcoendoplasmic reticulum Ca2+-ATPase
- STZ:
-
Streptozotocin
- TCA:
-
Tricarboxylic acid
- TRP:
-
Transient receptor potential
- TRPV1:
-
Transient receptor potential vanilloid 1
- VDCC:
-
Voltage-dependent Ca2+ channels
References
Voitenko NV, Kruglikov IA, Kostyuk EP, Kostyuk PG (2000) Effect of streptozotocin-induced diabetes on the activity of calcium channels in rat dorsal horn neurons. Neuroscience 95:519–524
Veves A, Backonja M, Malik RA (2008) Painful diabetic neuropathy: epidemiology, natural history, early diagnosis, and treatment options. Pain Med 9:660–674
Vincent AM, Callaghan BC, Smith AL, Feldman EL (2011) Diabetic neuropathy: cellular mechanisms as therapeutic targets. Nat Rev Neurol 7(10):573–583
Fernyhough P, Calcutt NA (2010) Abnormal calcium homeostasis in peripheral neuropathies. Cell Calcium 47(2):130–139
Verkhratsky A, Fernyhough P (2008) Mitochondrial malfunction and Ca2+ dyshomeostasis drive neuronal pathology in diabetes. Cell Calcium 44:112–122
Umeda M, Ohkubo T, Ono J, Fukuizumi T, Kitamura K (2006) Molecular and immunohistochemical studies in expression of voltage-dependent Ca2+ channels in dorsal root ganglia from streptozotocin-induced diabetic mice. Life Sci 79:1995–2000
Nazıroglu M (2009) Role of selenium on calcium signaling and oxidative stress-induced molecular pathways in epilepsy. Neurochem Res 34:2181–2191
Negi G, Kumar A, Sharma SS (2011) Melatonin modulates neuroinflammation and oxidative stress in experimental diabetic neuropathy: effects on NF-κB and Nrf2 cascades. J Pineal Res 50:124–131
Obrosova IG (2005) Increased sorbitol pathway activity generates oxidative stress in tissue sites for diabetic complications. Antioxid Redox Signal 7:1543–1552
Obrosova IG, Kador PF (2011) Aldose reductase/polyol inhibitors for diabetic retinopathy. Curr Pharm Biotechnol 12:373–385
Schmidt RE, Dorsey DA, Beaudet LN, Plurad SB, Parvin CA, Ohara S (2000) Effect of IGF-I and neurotrophin-3 on gracile neuroaxonal dystrophy in diabetic and aging rats. Brain Res 876:88–94
Pi J, Bai Y, Zhang Q, Wong V, Floering LM, Daniel K, Reece JM, Deeney JT, Andersen ME, Corkey BE, Collins S (2007) Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes 56:1783–1791
Sarre A, Gabrielli J, Vial G, Leverve XM, Assimacopoulos-Jeannet F (2012) Reactive oxygen species are produced at low glucose and contribute to the activation of AMPK in insulin-secreting cells. Free Radic Biol Med 52:142–150
Supale S, Li N, Brun T, Maechler P (2012) Mitochondrial dysfunction in pancreatic β cells. Trends Endocrinol Metab [Epub ahead of print]
Youn JY, Gao L, Cai H (2012) The p47(phox)- and NADPH oxidase organiser 1 (NOXO1)-dependent activation of NADPH oxidase 1 (NOX1) mediates endothelial nitric oxide synthase (eNOS) uncoupling and endothelial dysfunction in a streptozotocin-induced murine model of diabetes. Diabetologia 55:2069–2079
Aley KO, Levine JD (2002) Different peripheral mechanisms mediate enhanced nociception in metabolic/toxic and traumatic painful peripheral neuropathies in the rat. Neuroscience 111:389–397
Orreniu S, Nicotera P (1994) The calcium ion and cell death. J Neural Transm 43:1–11
Greene DA, Stevens MJ, Obrosova I, Feldman EL (1999) Glucose-induced oxidative stress and programmed cell death in diabetic neuropathy. Eur J Pharmacol 375:217–223
Li F, Obrosova IG, Abatan O, Tian D, Larkin D, Stuenkel EL, Stevens MJ (2005) Taurine replacement attenuates hyperalgesia and abnormal calcium signaling in sensory neurons of STZ-D rats. Am J Physiol Endocrinol Metab 288:29–36
Whyte KA, Greenfield SA (2002) Expression of voltage-dependent calcium channels in the embryonic rat midbrain. Brain Res Dev Brain Res 139:189–197
Wen XJ, Xu SY, Chen ZX, Yang CX, Liang H, Li H et al (2010) The roles of T type calcium channel in the development of neuropathic pain following chronic compression of rat dorsal root ganglia. Pharmacology 85:295–300
Li XY, Chen XG (2009) Role of PKCbeta in the malignant tumors and enzastaurin, a PKCbeta inhibitor. Yao Xue Xue Bao 44:449–455
Tahara M, Omatsu-Kanbe M, Sanada M, Maeda K, Koya D, Matsuura H, Kashiwagi A, Yasuda H (2006) Effect of protein kinase Cbeta inhibitor on Ca2+ homeostasis in diabetic sensory neurons. Neuroreport 17:683–688
Kostyuk E, Svichar N, Shishkin V, Kostyuk P (1999) Role of mitochondrial dysfunction in calcium signalling alterations in dorsal root ganglion neurons of mice with experimentally-induced diabetes. Neuroscience 90:535–541
Drel VR, Mashtalir N, Ilnytska O, Shin J, Li F, Lyzogubov VV, Obrosova IG (2006) The leptin-deficient (ob/ob) mouse: a new animal model of peripheral neuropathy of type 2 diabetes and obesity. Diabetes 55:3335–3343
Srinivasan K, Sharma SS (2011) Augmentation of endoplasmic reticulum stress in cerebral ischemia/reperfusion injury associated with comorbid type 2 diabetes. Neurol Res 33:858–865
Syed I, Kyathanahalli CN, Jayaram B, Govind S, Rhodes CJ, Kowluru RA, Kowluru A (2011) Increased phagocyte-like NADPH oxidase and ROS generation in type 2 diabetic ZDF rat and human islets: role of Rac1-JNK1/2 signaling pathway in mitochondrial dysregulation in the diabetic islet. Diabetes 60:2843–2852
Fernyhough P, Roy Chowdhury SK, Schmidt RE (2010) Mitochondrial stress and the pathogenesis of diabetic neuropathy. Expert Rev Endocrinol Metab 5:39–49
Adachi T (2010) Modulation of vascular sarco/endoplasmic reticulum calcium ATPase in cardiovascular pathophysiology. Adv Pharmacol 59:165–195
Kobayashi T, Taguchi K, Takenouchi Y, Matsumoto T, Kamata K (2007) Insulin-induced impairment via peroxynitrite production of endothelium-dependent relaxation and sarco/endoplasmic reticulum Ca(2+)-ATPase function in aortas from diabetic rats. Free Radic Biol Med 43:431–443
Turan B, Vassort G (2011) Ryanodine receptor: a new therapeutic target to control diabetic cardiomyopathy. Antioxid Redox Signal 15:1847–1861
Tian C, Shao CH, Moore CJ, Kutty S, Walseth T, DeSouza C, Bidasee KR (2011) Gain of function of cardiac ryanodine receptor in a rat model of type 1 diabetes. Cardiovasc Res 91:300–309
Yaras N, Tuncay E, Purali N, Sahinoglu B, Vassort G, Turan B (2007) Sex-related effects on diabetes-induced alterations in calcium release in the rat heart. Am J Physiol Heart Circ Physiol 293:H3584–H3592
Dror V, Kalynyak TB, Bychkivska Y, Frey MH, Tee M, Jeffrey KD, Nguyen V, Luciani DS, Johnson JD (2008) Glucose and endoplasmic reticulum calcium channels regulate HIF-1beta via presenilin in pancreatic beta-cells. J Biol Chem 283:9909–9916
Luciani DS, Gwiazda KS, Yang TL, Kalynyak TB, Bychkivska Y, Frey MH, Jeffrey KD, Sampaio AV, Underhill TM, Johnson JD (2009) Roles of IP3R and RyR Ca2+ channels in endoplasmic reticulum stress and beta-cell death. Diabetes 58:422–432
Nazıroğlu M (2007) New molecular mechanisms on the activation of TRPM2 channels by oxidative stress and ADP-ribose. Neurochem Res 32:1990–2001
Pieczenik SR, Neustadt J (2007) Mitochondrial dysfunction and molecular pathways of disease. Exp Mol Pathol 83:84–92
Özkaya D, Naziroğlu M, Armağan A, Demirel A, Köroglu BK, Çolakoğlu N, Kükner A, Sönmez TT (2011) Dietary vitamin C and E modulates oxidative stress induced-kidney and lens injury in diabetic aged male rats through modulating glucose homeostasis and antioxidant systems. Cell Biochem Funct 29:287–293
Kang J, Pervaiz S (2012) Mitochondria: redox metabolism and dysfunction. Biochem Res Int [Epub 2012 Apr 24]
Bravenboer B, Kappelle AC, Hamers FP, van Buren T, Erkelens DW, Gispen WH (1992) Potential use of glutathione fort he prevention and treatment of diabetic neuropathy in the streptozotocin—induced diabetic rat. Diabetologia 35:813–817
Sharma SS, Sayyed SG (2006) Effects of trolox on nerve dysfunction, thermal hyperalgesia and oxidative stress in experimental diabetic neuropathy. Clin Exp Pharmacol Physiol 33:1022–1028
Cameron NE, Cotter MA, Archibald V, Dines KC, Maxfield EK (1994) Anti-oxidant and pro-oxidant effects on nerve conduction velocity, endoneurial blood flow and oxygen tension in no-diabetic rats. Diabetelogia 37:449–459
Low PA, Nickander KK, Tritschler HJ (1997) The roles of oxidative stress and antioxidant treatment in experimental diabetic neuropathy. Diabetes 2:38–42
Nickander KK, Schmelzer JD, Rohwer DA, Low PA (1994) Effect of alpha-tocopherol deficiency on indices of oxidative stress in normal and diabetic peripheral nerve. J Neurol Sci 126:6–14
Kadiroglu AK, Sit D, Kayabasi H, Tuzcu AK, Tasdemir N, Yilmaz ME (2008) The effect of venlafaxine HCl on painful peripheral diabetic neuropathy in patients with type 2 diabetes mellitus. J Diab Compl 22:241–245
Yang T, Tsang KS, Poon WS, Ng HK (2009) Neurotrophism of bone marrow stromal cells to embryonic stem cells: noncontactinduction and transplantation to a mouse ischemic stroke model. Cell Transplant 18:391–404
Garrett NE, Malcangio M, Dewhurst M, Tomlinson DR (1997) alpha-Lipoic acid corrects neuropeptide deficits in diabetic rats via induction of trophic support. Neurosci Lett 222:191–194
Cameron NE, Jack AM, Cotter MA (2001) Effect of alpha-lipoic acid on vascular responses and nociception in diabetic rats. Free Radic Biol Med 31:125–135
Valsecchi AE, Franchi S, Panerai AE, Rossi A, Sacerdote P, Colleoni M (2011) The soy isoflavone genistein reverses oxidative and inflammatory state, neuropathic pain, neurotrophic and vasculature deficits in diabetes mouse model. Eur J Pharmacol 650(2–3):694–702
Oja SS, Saransaari P (2007) Pharmacology of taurine. Proc West Pharmacol Soc 50:8–15
Nazıroğlu M (2012) Molecular role of catalase on oxidative stress-induced Ca(2+) signaling and TRP cation channel activation in nervous system. J Recept Signal Transduct Res 32:134–141
Nazıroğlu M, Ozgül C (2012) Effects of antagonists and heat on TRPM8 channel currents in dorsal root ganglion neuron activated by nociceptive cold stress and menthol. Neurochem Res 37:314–320
Uchida K, Tominaga M (2011) The role of thermosensitive TRP (transient receptor potential) channels in insulin secretion. Endocr J 58:1021–1028
Uchida K, Dezaki K, Damdindorj B, Inada H, Shiuchi T, Mori Y, Yada T, Minokoshi Y, Tominaga M (2011) Lack of TRPM2 impaired insulin secretion and glucose metabolisms in mice. Diabetes 60:119–126
Romero JR, Castonguay AJ, Barton NS, Germer S, Martin M, Zee RY (2010) Gene variation of the transient receptor potential cation channel, subfamily M, members 6 (TRPM6) and 7 (TRPM7), and type 2 diabetes mellitus: a case-control study. Transl Res 156:235–241
Qian F, Huang P, Ma L, Kuznetsov A, Tamarina N, Philipson LH (2002) TRP genes: candidates for nonselective cation channels and store-operated channels in insulin-secreting cells. Diabetes 51(Suppl 1):183–189
Wei Z, Wang L, Han J, Song J, Yao L, Shao L, Sun Z, Zheng L (2009) Decreased expression of transient receptor potential impaires the postischemic recovery of diabetic mouse hearts. Circ J 73:1127–1132
Bishnoi M, Bosgraaf CA, Abooj M, Zhong L, Premkumar LS (2011) Streptozotocin-induced early thermal hyperalgesia is independent of glycemic state of rats: role of transient receptor potential vanilloid 1(TRPV1) and inflammatory mediators. Mol Pain 7:52
Pabbidi RM, Yu SQ, Peng S, Khardori R, Pauza ME, Premkumar LS (2008) Influence of TRPV1 on diabetes-induced alterations in thermal pain sensitivity. Mol Pain 4:9
Zsombok A, Bhaskaran MD, Gao H, Derbenev AV, Smith BN (2011) Functional plasticity of central TRPV1 receptors in brainstem dorsal vagal complex circuits of streptozotocin-treated hyperglycemic mice. J Neurosci 31:14024–14031
Wilder-Smith EP, Ong WY, Guo Y, Chow AW (2007) Epidermal transient receptor potential vanilloid 1 in idiopathic small nerve fibre disease, diabetic neuropathy and healthy human subjects. Histopathology 51:674–680
Ohanyan VA, Guarini G, Thodeti CK, Talasila PK, Raman P, Haney RM, Meszaros JG, Damron DS, Bratz IN (2011) Endothelin-mediated in vivo pressor responses following TRPV1 activation. Am J Physiol Heart Circ Physiol 301:1135–1142
Manni L, Rocco ML, Barbaro Paparo S, Guaragna M (2011) Electroacupuncture counteracts the development of thermal hyperalgesia and the alteration of nerve growth factor and sensory neuromodulators induced by streptozotocin in adult rats. Diabetologia 54:1900–1908
Ristoiu V, Shibasaki K, Uchida K, Zhou Y, Ton BH, Flonta ML, Tominaga M (2011) Hypoxia- induced sensitization of transient receptor potential vanilloid 1involves activation of hypoxia-inducible factor-1 alpha and PKC. Pain 152:936–945
Tanaka H, Shimaya A, Kiso T, Kuramochi T, Shimokawa T, Shibasaki M (2011) Enhanced insulin secretion and sensitization in diabetic mice on chronic treatment with a transient receptor potential vanilloid 1 antagonist. Life Sci 88:559–563
Liu XH, Qin C, Du JQ, Xu Y, Sun N, Tang JS, Li Q, Foreman RD (2010) Diabetic rats show reduced cardiac-somatic reflex evoked by intrapericardial capsaicin. Eur J Pharmacol 651:1–3
Talbot S, Chahmi E, Dias JP, Couture R (2010) Key role for spinal dorsal horn microglial kinin B1 receptor in early diabetic pain neuropathy. J Neuroinflamm 7:36
Mohammadi-Farani A, Sahebgharani M, Sepehrizadeh Z, Jaberi E, Ghazi-Khansari M (2010) Diabetic thermal hyperalgesia: role of TRPV1 and CB1 receptors of periaqueductal gray. Brain Res 1328:49–56
Kang JH, Goto T, Han IS, Kawada T, Kim YM, Yu R (2009) Dietary capsaicin reduces obesity- induced insulin resistance and hepatic steatosis in obese mice fed a high-fat diet. Obesity (Silver Spring) 18:780–787
Facer P, Casula MA, Smith GD, Benham CD, Chessell IP, Bountra C, Sinisi M, Birch R, Anand P (2007) Differential expression of the capsaicin receptor TRPV1 and related novel receptors TRPV3, TRPV4 and TRPM8 in normal human tissues and changes in traumatic and diabetic neuropathy. BMC Neurol 7:11
Wuensch T, Thilo F, Krueger K, Scholze A, Ristow M, Tepel M (2010) High glucose- induced oxidative stress increases transient receptor potential channel expression in human monocytes. Diabetes 59:844–849
Romero JR, Ridker PM, Zee RY (2009) Gene variation of the transient receptor potential cation channel, subfamily member 7 (TRPM7), and risk of incident ischemic stroke: prospective, nested, case-control study. Stroke 40:2965–2968
Song Y, Hsu YH, Niu T, Manson JE, Buring JE, Liu S (2009) Common genetic variants of the ion channel transient receptor potential membrane melastatin 6 and 7 (TRPM6 and TRPM7), magnesium intake, and risk of type 2 diabetes in women. BMC Med Genet 10:4
Landman N, Jeong SY, Shin SY, Voronov SV, Serban G, Kang MS, Park MK, Di Paolo G, Chung S, Kim TW (2006) Presenilin mutations linked to familial Alzheimer’s disease cause an imbalance in phosphatidylinositol 4,5-bisphosphate metabolism. Proc Natl Acad Sci USA 103:19524–19529
Mita M, Ito K, Taira K, Nakagawa J, Walsh MP, Shoji M (2010) Attenuation of store-operated Ca+2 entry and enhanced expression of TRPC channels in caudal artery smooth muscle from Type 2 diabetic Goto-Kakizaki rats. Clin Exp Pharmacol Physiol 37:670–678
Graham S, Ding M, Ding Y, Sours-Brothers S, Luchowski R, Gryczynski Z, Yorio T, Ma H, Ma R (2010) Abudance of TRPC6 protein in glomerular mesangial cells is decreased by ROS and PKC in diabetes. Am J Physiol Cell Physiol 201:304–315
Zbidi H, López JJ, Amor NB, Bartegi A, Salido GM, Rosado JA (2009) Enhanced expression of STIM/Orail and TRPC3 in platelets from patients with type 2 diyabetes mellitus. Blood Cells Mol Dis 43:211–213
Chung AW, Au Yeung K, Chum E, Okon EB, van Breemen C (2009) Diabetes modulates capacitative calcium entry and expression of transient receptor potential canonical channels in human saphenous vein. Eur J Phamacol 613:114–118
Liu D, Maier A, Scholze A, Rauch U, Boltzen U, Zhao Z, Zhu Z, Tepel M (2008) High glucose enhances transient receptor potential channel canonical type 6-dependent calcium influx in human platelets via phosphatidylinositol 3-kinase-dependent pathway. Arterioscler Thromb Vasc Biol 28:746–751
Niehof M, Borlak J (2008) HNF4 alpha and the Ca-channel TRPC1 are novel disease candidate genes in diabetic nephropathy. Diabetes 7:1069–1077
Li DP, Chen SR, Finnegan TF, Pan HL (2004) Signalling pathway of nitric oxide in synaptic GABA release in the rat paraventricular nucleus. J Physiol 554:100–110
Jagodic MM, Pathirathna S, Nelson MT, Mancuso S, Joksovic PM, Rosenberg ER, Bayliss DA, Jevtovic-Todorovic V, Todorovic SM (2007) Cell-specific alterations of T-type calcium current in painful diabetic neuropathy enhance excitability of sensory neurons. J Neurosci 27:3305–3316
Acknowledgments
There is no financial support and conflict interest in the current study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nazıroğlu, M., Dikici, D.M. & Dursun, Ş. Role of Oxidative Stress and Ca2+ Signaling on Molecular Pathways of Neuropathic Pain in Diabetes: Focus on TRP Channels. Neurochem Res 37, 2065–2075 (2012). https://doi.org/10.1007/s11064-012-0850-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-012-0850-x