
Abstract
Astrocytes, a type of glial cells in the brain, are eukaryotic cells, and a hallmark of these are subcellular organelles, such as secretory vesicles. In neurons vesicles play a key role in signaling. Upon a stimulus—an increase in cytosolic concentration of free Ca2+ ([Ca2+]i)—the membrane of vesicle fuses with the presynaptic plasma membrane, allowing the exit of neurotransmitters into the extracellular space and their diffusion to the postsynaptic receptors. For decades it was thought that such vesicle-based mechanisms of gliotransmitter release were not present in astrocytes. However, in the last 30 years experimental evidence showed that astrocytes are endowed with mechanisms for vesicle- and non-vesicle-based gliotransmitter release mechanisms. The aim of this review is to focus on exocytosis, which may play a role in gliotransmission and also in other forms of cell-to-cell communication, such as the delivery of transporters, ion channels and antigen presenting molecules to the cell surface.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The term “glia” was coined in the middle of nineteenth century by Rudolf Virchow for cells surrounding neurons. About 30 years ago it was shown that astrocytes, the most abundant glial cell type, exhibit a special form of cytoplasmic excitability (reviewed in [1, 2]). They respond to neurotransmitters, such as glutamate, by increasing intracellular Ca2+ concentration ([Ca2+]i), which was first measured by optical fluorescent probes [3]. This form of astrocytic excitability provides the ability to “sense” glutamatergic synaptic transmission [4]. This was followed by the discovery that astrocytic Ca2+ dynamics can yield astrocyte-neuron signaling, with at least two underlying mechanisms: direct, perhaps using gap junctions [5]; and indirect utilizing glutamate released from astrocytes via Ca2+-dependent exocytosis [6]. The later mode of signaling has led to the discovery of gliotransmission-based modulation of synaptic transmission [7]. However, the exact mechanisms of gliotransmitter release are a matter of debate among gliologists.
Several mechanisms appear to coexist in a single astrocyte (reviewed in [8, 9]). In addition to a number of nonvesicle-based mechanisms such as (i) channel opening induced by cell swelling, (ii) release through connexin hemichannels and pannexons on the cell surface and (iii) release through ionotropic purinergic receptors, (iv) through transporters, by means of reversal uptake by plasma membrane excitatory amino acid transporters, (v) by exchange via the cystine-glutamate antiporter and organic anion transporters, glial cells release chemical messengers also via (vi) the vesicle-based mechanisms. While all of these mechanisms require a concentration gradient along which chemical messengers are transported to their targets, the vesicle-based mechanisms have certain advantages over the non-vesicular modes of chemical messenger release. As the vesicular mode of gliotransmitter release is actively debated [10], it is appropriate to consider experimental evidence and key physical-biology concepts, supporting the notion that glial cells, as any other eukaryotic cell, may utilize vesicle-laden membrane storage organelles for signaling with neighboring cells.
Advantages of Vesicle- Versus Non-Vesicle-Based Transmitter Release
Vesicle-based transmitter release is mediated by exocytosis, involving the fusion between the vesicle and the plasma membranes. This universal process, an evolutionary invention of eukaryotic cells, emerged from a prokaryotic-like precursor cell by endosymbiosis [11]. Thus, a hallmark of eukaryotic cells, including astrocytes, are membrane-bound subcellular structures, including the nucleus, mitochondria, Golgi bodies and secretory vesicles. When eukaryotic cells evolved 1,000–2,000 million years ago [11, 12], this was associated with a cell volume increase by 3–4 orders in magnitude. The increased cell size dictated a new organizational make-up. A key reason for this is that signaling and communication within the relatively large eukaryotic cell volume could no longer be supported mainly by diffusion-based processes, which provide effective and rapid transport of molecules within the submicron range hence the development of subcellular organelles presented a solution for the “signaling problem” in the relatively large volume of eukaryotic cells. At least two important considerations support this notion.
First, relatively small sized subcellular structures (i.e. secretory vesicle), in which chemical messengers can be stored and concentrated, presents an advantage for “cell economy”. Transport of molecules across the membrane against the concentration gradient, such as l-glutamate entering a vesicle, consumes energy in the form of ATP hydrolysis. For example, it was estimated that the budget for recycling of 4,000 glutamate molecules into a vesicle, consists of 11,000 ATP molecules [13]. In contrast, the budget for concentrating glutamate into the cytosol of a whole cell must be much higher, since the volume of a vesicle (a sphere of 50 nm in diameter) is at least 9 orders of magnitude smaller than the volume of a typical cell (a sphere with a diameter of 15 μm). Therefore, a smaller (“more economical”) energy budget is required to attain a relatively high concentration of signaling molecules in the vesicle lumen versus that of the whole cytoplasm (Fig. 1).

Vesicle-and non-vesicle based mechanisms of transmitter release. a Astrocyte with transmitters (dots) accumulating in the cytosol. When these transmitters are concentrated in the cytosol, they may exit into the extracellular space via opening of the channels, for example. Since the establishment of a higher concentration of transmitter in the cytosol is energetically more “expensive” than the establishment of a higher concentration of transmitter within secretory vesicles (volume of the cytosol is orders of magnitude larger than the volume of a vesicle) it is more “economical” to use vesicles as means of transmitter release. Astrocytes have many mechanisms of non-vesicular secretion: (i) channel opening induced by cell-swelling, (ii) cysteine-glutamate antiporters and organic anion transporters, (iii) ionotropic purinergic receptors, (iv) connexin hemichannels/pannexons and (v) reversal of Na+-dependent excitatory amino acid transporters (EAAT). b This panel shows that vesicles containing transmitters can be positioned to various sites at the plasma membrane. Thus high concentration-loaded compartments are movable and can be strategically positioned where required for efficient signaling. The fusion of vesicles with the plasma membrane is mediated by proteins forming the SNARE complex
Second, secretory vesicles represent movable compartments, containing high concentrations of soluble signaling molecules. These compartments can be placed to sites within the cell, where a high concentration gradient of chemical messengers is required for efficient diffusional delivery to the targets. Vesicle compartments can be considered as “signal driving modules”, which can be strategically positioned within the cell. Such regulation cannot be attained with the machinery for the non-vesicular modes of chemical messenger release, unless traffic of these machineries involves targeted exocytosis.
Slow Regulated Exocytosis in Astrocytes
Vesicle-based release mechanisms have played a key role in the development of multicellular organisms, especially in cases where rapid communication between cells is required, such as that between neurons. For example, neurotransmitters are stored in secretory vesicles and can be discharged from them swiftly following a stimulus. By having these vesicles adjacent to the plasma membrane, in an active zone—special morphological feature in presynaptic terminals with clusters of secretory vesicles—the delay of signaling to the neighboring postsynaptic membrane is minimized and was measured to be as short as 100 μs [14]. On the other hand, the vesicle-based mechanisms of chemical messenger release can exhibit much longer delays, if the secretory vesicle delivery to the subcellular membrane signaling sites is attenuated and/or by regulating the vesicle cargo discharge through the fusion pore regulation [15]. The delays may be even several hours or days, if vesicles with cargo have to be synthesized following a signal application, as is the case in antigen presentation by astrocytes (see section on Exocytosis and regulation of plasma membrane signals) in pathological conditions.
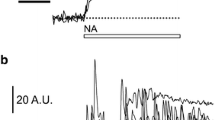
The properties of vesicle-based mechanisms of transmitter release can be studied at cellular level by monitoring changes in the plasma membrane area. The merger of the vesicle membrane with the plasma membrane contributes to changes in the plasma membrane area, which can be monitored directly by measuring membrane capacitance (Cm) [16], a parameter linearly related to the membrane area [17]. This technique was used in cultured astrocytes [18] to test the hypothesis that an increase in [Ca2+]i, elevated by photolysis of caged Ca2+ compounds, elicits an increase in the whole-cell Cm. Half-maximal response in Cm increase was attained at around 20 μM [Ca2+]i, which is similar to the Ca2+-dependency of regulated exocytosis in various types of neurons, recorded by a similar technique [16, 19, 20]. However, the kinetics of this response in astrocytes is at least two orders of magnitude slower in comparison to the kinetics of regulated exocytosis recorded by a similar technique in neurons [16] (Fig. 2). The relatively slow responsiveness of glial exocytosis may be due to many factors, including the slow delivery of vesicles to the plasma membrane fusion sites and/or due to distinct (slow) molecular mechanisms regulating the vesicle and the plasma membrane merger. Nonetheless, these results provided compelling evidence that in cultured astrocytes Ca2+-dependent regulated exocytosis is present. Moreover, the relatively slow regulated exocytosis in astrocytes indicates that astrocytes operate as signal integrators. However, which vesicles contribute to the plasma membrane area increases is unclear solely from measurements of Cm. In the next part of this section we consider evidence about the release of gliotransmitters from astrocytes.

Comparison of time-dependent changes in Cm recorded in a neuronal cell (a) and in an astrocyte (b). a Two types of Ca2+-induced increases in Cm in a photoreceptor have been recorded. Top trace was best fitted by a single exponential function (dotted line), while the bottom trace was best fitted to a sum of two exponential functions as shown by the equation below the horizontal line. The fastest rate constant (kf) was around 400 s−1 (modified from [16]). b Top trace shows time-dependent changes in [Ca2+]i, elicited by UV light flash photolysis of caged Ca2+ compound dialysed into the cytosol of the cell [18]. UV flash was applied at the time indicated by the arrow. Note that the rapid increase in [Ca2+]i following the UV flash application induced an exponential increase in Cm with a rate constant (k) of around 2 s−1 [18]. G denotes the real part of admittance trace
Astrocytic Transmitters
Essential elements of triggered release of astrocytic transmitters are mechanisms of Ca2+ delivery. In neurons positioning of Ca2+ sources (mainly voltage-dependent channels) to the proximity of exocytotic machinery minimizes the latency in stimulus-secretion coupling [21, 22]. In astrocytes several sources of Ca2+ have been described (reviewed in [1]). Briefly, there are three main mechanisms that enable increases in [Ca2+]i in astrocytes, which involve three different compartments: endoplasmic reticulum (ER), mitochondria, and extracellular space. ER has machinery that enables dynamic Ca2+ storage and generation of cytosolic Ca2+ signals. Ca2+ ATPases pump Ca2+ into ER, providing steep concentration gradient between ER lumen and cytosol. Physiological stimulation can cause opening of specific channels: inositol 1, 4, 5-triphosphate receptors (IP3) [23] and ryanodine receptors, which need to operate jointly to generate Ca2+-dependent glutamate release [24]. Second, mitochondria are able to retain Ca2+ by accumulating significant amounts of Ca2+ through highly selective mitochondrial Ca2+ uniporters or release Ca2+ through mitochondrial permeability transition pore or Na+/Ca2+ exchangers. Finally, plasma membrane channels and receptors additionally contribute as Ca2+ sources. Voltage-gated Ca2+ channels are functionally expressed in at least cultured astrocytes [25], likely causing Ca2+ cytosolic increases upon strong depolarization with subsequent glutamate release [26]. Additionally, they are found in immature astrocytes in situ [27], but their presence in mature astrocytes remains unknown. The store-operated entry upon depletion of the ER stores is likely governed by canonical transient receptor potential (TRPC) proteins, forming heteromultimeric channels composed of TRPC1, TRPC4 and TRPC5 [28, 29]. A new more sensitive, genetically encoded Ca2+ sensor, such as GCaMP3, revealed that TRPA1 contributes to the regulation of resting [Ca2+]i [30]. Ionotropic Ca2+ permeable receptors (reviewed in [31]) represented by AMPA/NMDA glutamate and P2X purinoreceptors can also contribute to the Ca2+ influx. Additionally, Na+/Ca2+ exchangers can contribute to Ca2+ entry to the cytosol when operating in reverse mode [32, 33]. Taken together, various sources operate dynamically to generate oscillations in [Ca2+]i which can subsequently lead to the Ca2+-dependent transmitter release.
In addition to the essential role of Ca2+-triggering mechanisms, for a transmitter to be qualified as a gliotransmitter, other criteria have to be fulfilled as well. As for the neurotransmitters [34], to classify a molecule released from glia/astrocyte as a “gliotransmitter” [9, 35–37] the following criteria must be considered: (i) synthesis and/or storage in glia; (ii) regulated release triggered by physiological and/or pathological stimuli; (iii) activation of paracrine or autocrine responses; and (iv) a role in (patho)physiological processes. In the next sections we address gliotransmitters such as: amino acids (glutamate, GABA and d-serine); peptides, like atrial natriuretic peptide (ANP) and nucleotides, such as adenosine 5′-triphosphate (ATP).
Amino Acids as Astrocytic Transmitters
In astrocytes, glutamate is synthesized de novo [38], as a by-product of the tricarboxylic acid (TCA) cycle, involving the astrocyte-specific enzyme pyruvate carboxylase. Glutamate is converted from the TCA cycle intermediate, α-ketoglutarate, usually via transamination of another amino acid, such as, aspartate [39]. All the three known isoforms of vesicular glutamate transporters (VGLUTs) 1, 2, and 3, which use the proton gradient created by vacuolar type H+ ATPases (V-ATPases) to package glutamate into vesicles, have been detected in astrocytes [18, 40–45].
d-serine is converted from l-serine by the action of serine racemase, an enzyme found in astrocytes [46, 47] and also abundantly in neurons [47–49]. Astrocytes are thought to serve as the key metabolic provider of l-serine which is shuttled to neurons, likely via membrane transporters, for d-serine production in neurons [48]. In addition to the non-vesicle-based mechanisms of release, astrocytes release d-serine by regulated exocytosis in an activity dependent manner [50, 51]. How d-serine is loaded into astrocytic vesicles is not clear yet, perhaps via a vesicular d-serine transporter.
Astrocytes can accumulate, synthesize and release GABA as summarized [52, 53]. Astrocytes have the capacity to accumulate GABA as shown with anti-GABA antibodies and express transporters for GABA. GABA can be produced by two distinct pathways, namely by conversion of glutamate to GABA by the enzyme glutamic acid decarboxylase (GAD) or by the monoacetylation of putrescine. The GAD expression in glial cells, however, is significantly lower than in neurons. Production of GABA from putrescin in astrocytes is low, but is upregulated under pathological conditions. Astrocytes chronically release GABA likely through a novel anion channel, bestrophin-1 and thereby mediate tonic inhibition. Bestrophine-1 can be modulated by changes in intracellular Ca2+ and cell volume, but is even tonically active at resting Ca2+ levels [54]. In addition to this non-vesicular mechanism of release, there is evidence that the vesicular inhibitory amino acid transporter (VIAAT) is expressed in astrocytes as well [55]. Thus besides GABA being localized in the cytosol from where it may be released into the extracellular space via nonvesicle-based mechanisms, it may also be present in vesicles. Recent study has provided evidence for release of GABA through Na+-dependent GABA transporter GAT, which could contribute to accelerated glutamate uptake via EAATs [56]. While, unequivocal evidence to support regulated exocytosis of GABA from astrocytes is yet to be provided, the vesicle-based mechanisms of other gliotransmitters, such as l-glutamate, ANP, and ATP are more extensively covered in the literature [1, 9].
In the first experiments where Ca2+-dependent glutamate release from cultured astrocytes was measured, high performance liquid chromatography was used [6]. Addition of ionomycin to increase cytosolic Ca2+ activity caused an increase in the release of glutamate, but only if external Ca2+ was present in the bathing medium of astrocytes. These data are consistent with the view that elevated [Ca2+]i is sufficient and necessary to stimulate glutamate release. Other stimuli that increase astrocytic [Ca2+]i, such as mechanical stimulation [6, 7, 24, 40, 57, 58], photostimulation [6], and photolysis of Ca2+ cages [43, 57, 59], all evoked release of glutamate.
Vesicle-based, Ca2+-dependent release of transmitters depends on the presence of exocytotic secretory machinery [60]. As neurons, astrocytes express proteins of the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex: synaptobrevin 2 ((Sb2), also referred to as vesicle–associated membrane protein 2 [61]), syntaxin 1, synaptosome-associated protein of 23 kDa (SNAP-23), as well as several ancillary proteins to this complex, including synaptotagmin 4 (see [1, 62, 63]). The use of Clostridial, tetanus, and various types of botulium toxins, which cleave exocytotic SNARE proteins, caused in astrocytes a reduction of Ca2+-dependent glutamate release (reviewed in [62]), a reduction in the UV flash-induced Cm increase [18], a reduction in the number of amperometric spikes [64], and a reduction in the glutamate-evoked sniffer-cell currents [58]. Similarly, the expression of mutated synaptotagmin 4, acting in a dominant-negative manner, caused the reduction of Ca2+-dependent glutamate release from astrocytes [65].
The morphology of astrocytic secretory vesicles, essential elements for regulated Ca2+-dependent exocytosis, has been investigated (Table 1). Based on immuno-electron microscopy (IEM), Sb2 can be found located in the vicinity of electron-lucent (clear) vesicular structures [66]. Sb2-positive vesicles were shown to be heterogeneous in size, ranging from 30 to over 100 nm in diameter [45], VGLUTs 1 or 2 in astrocytes in situ were found associated with small, clear vesicles with a mean diameter of ~30 nm [44]. Recycling glutamatergic vesicles which can capture the extracellular antibody against VGLUT1 in a Ca2+-dependent manner are electron-lucent and have a diameter of ~50 nm [67]. By using immunogold EM it was found that l-glutamate and d-serine accumulate in the synaptic like microvesicles with a diameter of 33 nm in fixed hippocampal perisynaptic astrocytes [68]. Much larger vesicles, over 1,000 nm in diameter, have been observed to form within minutes of repeated stimulation with pharmacological dosages (5–50 mM) of glutamate [69, 70]; these vesicles can release glutamate, although it is highly likely that they represent a pharmacologically-induced phenomenon or may play a role in pathological processes (see also the discussion in [71]).
The recycling of secretory vesicles at the plasma membrane has been investigated in astrocytes using fluorescence microscopy. Application of ionomycin in the presence of extracellular Ca2+, but not in its absence, caused uptake of the membrane recycling dye, FM 4-64 [72]. Similarly, using a pre-loading technique that stimulated membrane recycling and the trapping of styryl dyes (FM 1-43 or FM 2-10) in secretory organelles, astrocytes displayed a punctate pattern of FM fluorescence [58, 64]. Trafficking of glutamatergic vesicles in astrocytes was assessed using an immunological approach. After increasing cytosolic Ca2+ levels in astrocytes in the presence of extracellular antibodies against VGLUT1, presumably binding to luminal/intravesicular epitope of this transporter, there was an increase in intracellular fluorescent puncta [67]. The delivery of secretory vesicles and fusion to the plasma membrane was also studied in astrocytes. Crippa et al. [45] expressed a chimeric protein, where enhanced green fluorescent protein (EGFP) was fused to the vesicle luminal C-terminus of Sb2 (Sb2-EGFP), in astrocytes. Stimulation with Ca2+ ionophore resulted in the reduction of fluorescent Sb2-EGFP puncta, but with a simultaneous increase in plasma membrane fluorescence, consistent with regulated exocytosis and fusion of labeled vesicles to the plasma membrane. Net addition of vesicular membrane to the plasma membrane, monitored as changes in Cm, resulted in an increase of Cm, while concomitant measurements recorded a release of glutamate [43].
Experiments using total internal reflection fluorescence (TIRF) microscopy provided further evidence for vesicular exocytosis from astrocytes [44, 58, 73–75], where exocytosis of VGLUT1-, VGLUT2- or Sb2- positive vesicles were reported. As a consequence of vesicular fusions, quantal events of glutamate release, representing an exocytotic hallmark [76], have been recorded from astrocytes. Such events were detected using reporter “sniffer” cells expressing glutamate receptors [58, 77], or by amperometric measurements used to detect the release of dopamine, acting as a “surrogate” transmitter for glutamate, from glutamatergic vesicles [64].
Astrocytes can also release the amino acid d-serine [78], a ligand to the glycine modulatory binding site of the NMDA receptor. Mothet et al. [79] investigated the mechanism of this release using an enzyme-linked assay to measure extracellular d-serine concentration and established that astrocytes released d-serine in a Ca2+-dependent manner; the release was augmented by Ca2+ ionophore and inhibited by removal of extracellular Ca2+. Furthermore, the release of d-serine was reduced by concanamycin A, a V-ATPase inhibitor, and tetanus toxin, implicating the involvement of a vesicular mechanism. Consistent with this notion, d-serine was found to co-localize with Sb2 based on immunocytochemistry and fluorescence microscopy. The investigation of the mechanism underlying a Ca2+-dependent release of d-serine from astrocytes was expanded [80] by using pharmacological inhibition of vesicular budding, which revealed that d-serine was packaged in vesicles downstream of the Golgi apparatus. It is likely that d-serine is localized in glutamatergic vesicles, as studied by immunogold detection of l-glutamate and d-serine in hippocampal astrocytes [68]. While the results point to the d-serine being secreted from astrocytes also by regulated exocytosis, recent results of abundant expression of serine racemase also in neurons reveals a more complex picture of how astrocytes utilize d-serine in glial-neural communication [48].
Peptides as Astrocytic Transmitters
Unlike amino acids, which can get loaded into vesicles via membrane transporters, peptidergic messengers enter vesicles via the synthetic secretory pathway. These peptides are typically made as pro-peptides in the ER, transit Golgi compartments where they get concentrated and sorted into organelles; then, they are processed in vesicles to their final form before release [81]. Classical view holds that vesicles carrying peptidergic transmitters appear to have a distinct morphological appearance under electron microscopy [69]. They have relatively larger diameters in comparison to the synaptic-like vesicles, they exhibit electron dense cores, thus they are termed dense-core vesicle, large dense-core vesicles or secretory granules. Dense-core vesicles with diameters of around 100 nm, containing secretogranin II (SgII) were reported in astrocytes [82]. Furthermore, it was shown that SgII is released upon stimulation by different secretagogues, including bradykinin, 3′,5′- cyclic adenosine monophosphate (cAMP), phorbol 12-myristate 13-acetate (PMA) and the Ca2+ ionophore ionomycin, which in combination with PMA appeared to be the most effective stimulus for SgII release from astrocytes [82]. This study also reported that astrocytes contain fewer smaller and less dense secretory granules containing SgII, indicating that peptidergic vesicles in astrocytes are not uniform in morphological appearance. Neuropetide Y was shown to be contained in subcellular compartments distinct from synaptic like-vesicles in astrocytes [83].
One of the first messenger peptides studied for exocytotic release from astrocytes was ANP. The function of ANP release from astrocytes, may play a role in cerebral blood regulation [84] and also as an autocrine control of astrocyte function, since all the natriuretic peptide receptors are expressed by astrocytes (see [85, 86]). To study the discharge of ANP, Krzan et al. [72] transfected astrocytes with a construct to express pro-ANP fused with the emerald green fluorescent protein (ANP.emd). The number of fluorescent ANP.emd puncta was reduced upon stimulating the cells by ionomycin and was strictly depended on the extracellular Ca2+. Concomitant with the Ca2+-dependent decrease in fluorescent puncta, the fluorescence intensity of the FM 4-64 dye, a reporter of cumulative exocytosis, increased in a Ca2+-dependent manner as well. Together these data strongly indicated that regulated exocytosis mediates the release of ANP from astrocytes. Interestingly, vesicles containing ANP also appear to contain ATP [87], which is consistent with the report that ATP is stored in SgII-containing vesicles [88]. Thus, given that ATP and peptides have different molecular sizes it is possible that differential release of these two chemical messengers from a single vesicle may be attained by the regulation at the single vesicle fusion-pore level [89].
In atrial myocytes, EM shows that pro-ANP is condensed in the trans-Golgi network and, because pro-ANP is cleaved only on release, secretory vesicles budding off the trans-Golgi network are already mature. Their shape, size (120–175 nm) and electron density appears to be heterogeneous, determined by the aggregation of the pro-ANP in vesicles [90]. In astrocytes the size of ANP recycling vesicles was studied by IEM after exposing live astrocytes to extracellular anti-ANP antibody, which sequestered within vesicles with diameters ranging between 30 to 100 nm [91]. The mobility of these recycling ANP-containing vesicles was one order of magnitude smaller than that of ANP-containing vesicles trafficking to the plasma membrane vesicle-docking site [92, 93].
The mobility of anti-ANP antibody capturing vesicles and endosomes/lysosomes is dramatically reduced upon the stimulation of cells [91, 94], which differs from the stimulation-increased mobility of anti-VGLUT1 antibody capturing vesicles in astrocytes [67]. Clearly, this indicates that vesicles with different chemical messenger content may traffic within the cytoplasm of astrocytes by distinct mechanisms. However, functional significance of these observations is not clear, but the results clearly show that the mobility of vesicles retrieving from the plasma membrane is subject to the (patho)physiological state of the astrocyte [91, 94–96]. This may play a role in the regulation of the vesicle cycle and vesicle cargo discharge [91]. Furthermore, recycling vesicles may not only carry luminal cargo but, also, membrane associated signaling molecules which participate in cell-to-cell contact interactions [97, 98] or in determining the density of plasma membrane transporters [99], such as, the glutamate transporter EAAT2 [100].
Astrocytes also contain recycling vesicles, specialized endocytic compartments, which may serve for bidirectional communication between neurons and glia. On one hand, these vesicles may take-up extracellular peptides, process them, and recycle them back into the extracellular space via secretory pathway and regulated exocytosis. When studying the activity-dependent secretion of BDNF and its extracellular availability, Bergami et al. [101] conducted interesting experiments and provided evidence that BDNF, which is de novo synthetized in neurons, gets secreted after theta-burst stimulation in its pro-form into the extracellular medium. Then the pro-BDNF is rapidly internalized by endocytosis (BDNF vesicle diameter was 125 nm) into astrocytes and then recycled and their content secreted upon stimulation. Taken together, this study shows that endocytic vesicles may represent a storage/recycling compartment for endocytosed pro-BDNF before routing it to the SNARE-dependent secretory pathway [101].
In summary peptidergic vesicles in astrocytes are heterogeneous in nature and may, as is the case of SgII- and ANP-containing vesicles, also contain ATP [87, 88].
ATP as an Astrocytic Transmitter
Intracellular ATP provides energy for a variety of processes, including vesicular recycling and is produced via glycolysis and oxidative phosphorylation to reach cytoplasmic concentrations of over 10 mM, thus establishing a concentration gradient favouring ATP exit from cells. Once released into extracellular space, ATP can be used in intercellular signaling acting directly onto purinergic receptors. Alternatively, upon its hydrolysis by membrane-bound ecto-nucleotidases, the extracellular degradation products, ADP and adenosine, can activate different plasma membrane receptors (reviewed in [102, 103]).
In addition to the cytoplasmic localization of ATP, astrocytes possess secretory vesicles in which ATP appears to be concentrated together with the secretory peptide SgII [82, 88]. The subcellular fractionation experiments revealed that SgII structures were mainly distinct from fractions containing Sb2 [82], consistent with the finding that dense core vesicles represented 1–2 % of the total number of immuno-isolated Sb2-containing vesicles [45] and with the confirmation that a very low abundance of dense-core granules is present in astrocytes [104]. Similarly, using IEM, it was demonstrated that Sb2 can be associated with some dense core vesicles, with diameters ranging from 100 from 700 nm [66]. Following subcellular fractionations, immunoblotting for several exocytotic proteins, Sb2, syntaxin 1, cellubrevin and synaptotagmin 1, were found to co-localize with ATP containing organelles [66]. It should be noted, however, that the presence of synaptotagmin 1 was not detected in astrocytes by others [6, 45, 65]. The protein responsible for the ATP accumulation in secretory vesicles has recently been identified as SLC17A9 [105] a vesicular nucleotide transporter (VNUT) that also appears to be present in astrocytes [106].
Morphological and biochemical evidence suggests that ATP as an astrocytic chemical messenger may be released by Ca2+-dependent exocytosis. The first evidence in support of such a notion comes from experiments in which astrocytes exposed to nitric oxide responded with an increase in cytoplasmic Ca2+ and the release of ATP to the extracellular space [107]. Buffering of intracellular Ca2+ with BAPTA or preventing vesicular release with botulinum toxin C greatly reduced the release of ATP. Furthermore, Coco et al. [88] demonstrated that mechanically stimulated astrocytes released ATP which could be inhibited by application of bafilomycin A1 or tetanus toxin. Stimulation of cultured astrocytes could be evoked by uridine 5′-triphosphate (UTP) via the likely activation of P2Y2 receptors [108] and was reduced by thapsigargin and lithium ions that can block the intracellular generation of IP3. It is likely that the exocytotic pathway is involved in UTP-induced ATP release from astrocytes since brefeldin A, a blocker of transport vesicles budding off the Golgi apparatus, cytochalasin D, a disruptor of actin microfilaments, and botulinum toxin A, the exocytosis inhibitor, all blocked ATP release [108].
To study the quantal nature of ATP release from astrocytes, Pangrsic et al. [87] incubated astrocytes with quinacrine, a compound that fluorescently labels ATP containing structures. Using TIRF microscopy, quinacrine showed punctate stain. The rapid loss of these puncta was evident upon receptor stimulation using glutamate or ATP and stimuli that directly raise intracellular Ca2+ levels, ionomycin or flash photolysis of caged Ca2+ [109]. Expressing a dominant negative SNARE (dnSNARE) in astrocytes resulted in the inhibition of the Ca2+-induced reduction in the quinacrine fluorescent puncta representing ATP-containing vesicle exocytosis [87]. Glutamate stimulation of astrocytes showed quantal release of ATP as recorded by ATP reporter cells [87], human embryonic kidney cells expressing a mutated P2X3 receptor with reduced desensitization. A similar approach may be used to study quantal ATP release from brain slices, however, discrimination of whether ATP is released from non-astrocytes cannot be controlled as well as from in vitro experiments. Experiments with mice expressing dnSNARE in astrocytes only [110] additionally led to the discovery that ATP is a major gliotransmitter in vivo, which can be converted to adenosine in extracellular space under physiological conditions, affecting tonic A1 receptor-mediated presynaptic inhibition of excitatory synaptic transmission.
Interestingly the exocytotic release of ATP stored in astrocytic lysosomes could be detected only with unphysiologically long stimulation [111], exhibiting different sensitivity in comparison to other types of vesicles [58], indicating that distinct exocytotic mechanisms control release from vesicle subtypes. Consistent with this, only lysosomes carrying the VAMP7/TI-VAMP appear to be fusion competent [112]. Distinct vesicle fusion mechanisms are consistent with distinct mobility properties of astrocytic vesicles, which are, in part, determined by intermediate filament cytoskeleton [94, 95] and by other molecules, such as monomeric GTPases [113]. One of the questions to be studied in the future will be how these molecular entities respond to changes in cytosolic Ca2+ to exert changes in vesicle movements.
Exocytosis and Regulation of Plasma Membrane Signaling
Exocytosis of astrocytic vesicles may not only serve for luminal cargo release but also for the delivery of plasma membrane associated receptors and transporters, such as major the histocompatibility complex (MHC) class II molecules [Vardjan et al., in preparation] and sodium-dependent glutamate type 1 transporters (GLT-1) [100].
MHC class II molecules are expressed on the surface of astrocytes only upon exposure to the pro-inflammatory cytokine interferon-gamma (IFN-γ). IFN-γ-activated astrocytes participate in antigen presentation and activation of CD4 helper T-cells in immune-mediated central nervous system disorders [114]. In antigen presenting cells (B-cells, dendritic cells, macrophages) MHC class II molecules reach the cell surface in membrane-bound vesicles. Our recent study showed [Vardjan et al., in preparation] that 48 h treatment of astrocytes with IFN-γ induces the expression of MHC class II molecules on the plasma membrane and in punctuate structures (Fig. 3) identified as late endosomes/lysosomes, which could be specifically labeled with Alexa Fluor546-conjugated dextran. Upregulation of intermediate filaments (IFs) in astrocytes is the hallmark of reactive gliosis [115] and has been observed also in IFN-γ-activated astrocytes [116]. Recent results have revealed that IFs are important for the regulation of vesicle traffic in astrocytes [95], however whether the mobility of vesicles involved in antigen presentation is affected by conditions of altered expression of IFs is unclear.

MHC class II–positive compartments appear in astrocytes 48 h following the addition of IFN-γ. Fluorescence images of control (Ctrl.) and IFN-γ-treated (IFN-γ) cultured mouse astrocytes labeled with fluorescein-tagged antibodies against MHC class II molecules (MHCII). IFN-γ induces punctuate expression of MHC class II molecules. TL, transmitted light. Scale bars: 10 μm
Glutamate is removed from the synaptic cleft by a family of glutamate transporters (GLTs), which are integral membrane proteins essential for maintaining physiological levels of glutamate. GLT-1 (EAAT2), which is localized almost exclusively on astrocyte processes surrounding the synapse, represents the predominant rout for clearances of glutamate in the CNS. Functional inactivation of GLT-1 may raise extracellular glutamate to toxic levels causing glutamate-mediated excitotoxicity. Using GFP-tagged GLT-1 Zhou and Sutherland [117] have shown that in astrocytes GLT-1 is expressed on plasma membrane and in clusters, which are intracellular structures that colocalize with early and recycling endosome and lysosome markers [118]. Treatment with PKC activator PMA reduces the level of GFP-GLT-1 molecules on the plasma membrane and increases the number of GLT-1 clusters. Cotransfection of cell by the dominant negative form of dynamin prevents PMA–induced GLT internalization and cluster formation. This indicates that GLT-1 traffics between plasma membrane and endosome/lysosome structures via a PKC-dependent clathrin mediated endocytic/recycling pathway [117]. In another study, it was shown that Ca2+-ionophore ionomycin triggers local increases and decreases in plasma membrane levels of GFP-GLT-1, which were not observed in the absence of extracellular Ca2+ [100]. The results suggest that besides PKC dependent endocytic/recycling pathway [117], Ca2+-dependent endocytosis/exocytosis may play important role in the regulation of membrane surface levels of GLT-1 in astrocytes.
Astrocytes express various G-protein coupled receptors (GPCRs) on their surface, which respond to signaling molecules released by neurons and/or astrocytes, including cannabinoid receptor 1 [119, 120], chemokine receptor CXCR4 [121], and P2Y1R receptor [74]. Activation of GPCRs on astrocytes is involved in regulation of exocytotic release of gliotransmitters such as glutamate from astrocytes via signaling cascades resulting in elevations in [Ca2+]i from internal Ca2+-stores. Glutamate released from astrocytes may stimulate metabotropic glutamate receptors on nearby neurons.
CBR1 receptors upon activation with endocannabinoides released from neurons trigger PLC-dependent [Ca2+]i increase from internal stores and subsequent glutamate release from astrocytes [120]. CBR1 is mainly expressed in intracellular acidic organelles, which colocalize with endocytic compartments. Trafficking of CBR1 compartments has been recently studied using CBR1 chimeras [122], however the mechanism by which CBR1 reach the surface of astrocytes, either by a constitutive recycling pathway or by a Ca2+-dependent mechanisms such as exocytosis, needs to be determined.
Activation of GPCR receptor CXCR4 [121] on astrocytes by chemokine stromal cell-derived factor-1 [123] or by human immunodeficiency virus coat glycoprotein 120 [121] triggers release of pro-inflammatory cytokine tumor necrosis factor alpha (TNF-α) from astrocytes. TNF-α acts in an autocrine fashion by triggering regulated exocytotic release of glutamate from the same or neighboring astrocytes. This happens with a relatively fast time-scale in the order of few hundred milliseconds [121], taking into account that astrocytes are electrically non-excitable and thus exocytosis relies only on a signaling pathway. The latter involves TNF-α binding to a TNF receptor 1 on astrocytes, which is followed by the production and release of prostaglandin E2 (PGE2), binding of PGE2 to its receptor that leads to an increase in [Ca2+]i from internal Ca2+-stores, and finally exocytosis of glutamate from astrocytes [123, 124]. TNF-α is released from astrocytes also upon binding of ATP to GPCR receptor P2Y1R [74]. Glial derived TNF-α can act also in a paracrine fashion. It can mediate AMPA and GABAA receptor trafficking in neurons by increasing exocytosis of AMPA receptors and endocytosis of GABAA receptors. An increase in surface levels of AMPA receptors and a decrease in GABAA receptors lead to strengthening of excitatory synapses or weakening of inhibitory ones [125]. Trafficking of CXCR4 and P2Y1R between plasma membrane and intracellular endocytic compartments in astrocytes has not been studied yet and may have important regulatory role in determining the availability of these receptors on the plasma membrane. Understanding of precise regulation of TNF-α-induced glutamate exocytosis via CXCR4 or P2Y1R receptor-mediated pathway is essential for the control of synaptic efficacy and any disregulation may contribute to glutamate excitotoxicity.
Future Directions
Astrocytes, as any eukaryotic cell, contain secretory vesicles. These compulsory morphological elements for exocytosis exhibit a large variety of diameters in astrocytes (reviewed in [9, 62, 126]). Future experiments will have to advance the current findings that Sb2 can be associated with vesicular structures ranging from 30 to 700 nm, the majority of which are electron-lucent (clear), while a small fraction (<2 %) are dense and not so dense [45, 66, 82, 88, 104]. Whether the larger secretory vesicles (~300 nm), shown to be engaged in quantal exocytotic release [64], represent endosome/lysosomes is yet to be investigated. Is the nature of fusion of these structures with the plasma membrane similar? What is the nature of vesicles bringing receptors, transporters and other signaling molecules to the cell surface?
Often the question is asked whether gliotransmitter release from astrocytes occurs in vivo under normal conditions? Special model animals have been developed to study the role of astrocytic regulated exocytosis in animal behavior, such as the dnSNARE mouse, expressing a cytosolic domain of Sb2 in astrocytes in the adult brain due to conditional and selective transgenic targeting. Initial studies in situ using hippocampal slices revealed that the expression of dnSNARE in astrocytes led to an increase in the magnitude of excitatory synaptic transmission at the CA3-CA1 synapse [127]. By performing pharmacological studies it was determined that this enhancement of synaptic transmission resulted from a reduction in the activation of presynaptic A1 receptors [127]. It is well known that there is a basal level of extracellular adenosine that tonically exerts a presynaptic inhibition of synaptic transmission. Consequently, these results led to conclusion that astrocytic dnSNARE expression removed the source of adenosine, thereby enhancing synaptic transmission [127]. Clearly, to carry out experiments in tissue slices or even in the whole brain to demonstrate directly the involvement of regulated exocytosis in gliotransmission with the space–time resolution available in experiments on single cells, is difficult, if not impossible, since glutamate and ATP are rather common metabolites and identifying the subcellular source and mechanism of release in vivo would be technically most challenging. Finally, one should take this into account when addressing the problem of comparing results obtained in cell culture versus in vivo.
In neurons and chromaffin cells as models, two modes of vesicle fusion have been described: (i) full fusion, where the vesicle collapses into the plasma membrane upon fusion, and (ii) transient fusion, where vesicles remain associated with the plasma membrane and transiently open the fusion pore. While both modes of exocytosis appear to occur in astrocytes [44, 64, 73, 75, 128, 129], it is unclear whether larger or smaller vesicles prefer one or the other mechanism. Single vesicle fusion studies have shown that diameter determines the fusion-pore properties [130] and that Munc18-1, a SNARE-interacting protein, also present in astrocytes, affects the fusion pore physiology [131]. The consequence of transient versus full fusion on release of gliotransmitters critically depends on the measurements of the fusion pore. Interestingly, even if a vesicle contains different types of gliotransmitters (i.e., peptide vs. nucleotide), these could be released differentially during transient fusion events [15, 132]. In turn, such differential release properties of the two forms of vesicular release could have profound differential effects on astrocytic modulation of synaptic transmission and plasticity at the tripartite synapse. On the other hand, it is also critical to understand the nature of vesicle fusion in cases where vesicles deliver molecules to the plasma membrane, such as those involved in the presentation of antigens by astrocytes under pathological conditions. Finally, the fastest response of regulated exocytosis in astrocytes is much slower than in neurons. Thus astrocytes are elements for relatively slow time-domain communication and act as signal integrators in the brain networks.
References
Parpura V, Verkhratsky A (2011) The astrocyte excitability brief: from receptors to gliotransmission. Neurochem Int. doi:10.1016/j.neuint.2011.12.001
Verkhratsky A et al (2011) Where the thoughts dwell: the physiology of neural-glial “diffuse neural net”. Brain Res Rev 66:133–155
Cornell-Bell AH et al (1990) Glutamate induces calcium waves in cultured astrocytes: long-range glial signaling. Science 247:470–473
Dani JW et al (1992) Neuronal activity triggers calcium waves in hippocampal astrocyte networks. Neuron 8:429–440
Nedergaard M (1994) Direct signaling from astrocytes to neurons in cultures of mammalian brain cells. Science 263:1768–1771
Parpura V et al (1994) Glutamate-mediated astrocyte-neuron signalling. Nature 369:744–747
Araque A et al (1998) Glutamate-dependent astrocyte modulation of synaptic transmission between cultured hippocampal neurons. Eur J Neurosci 10:2129–2142
Domingues AM et al (2010) Glia as transmitter sources and sensors in health and disease. Neurochem Int 57:359–366
Parpura V, Zorec R (2010) Gliotransmission: exocytotic release from astrocytes. Brain Res Rev 63:83–92
Smith K (2010) Neuroscience: settling the great glia debate. Nature 468:160–162
Cooper GM (2000) The origin and evolution of cells. Sunderland, Massachusetts
Yoon HS et al (2004) A molecular timeline for the origin of photosynthetic eukaryotes. Mol Biol Evol 21:809–818
Attwell D, Laughlin SB (2001) An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab 21:1133–1145
Sabatini BL, Regehr WG (1999) Timing of synaptic transmission. Annu Rev Physiol 61:521–542
Vardjan N et al (2009) The fusion pore and vesicle cargo discharge modulation. Ann N Y Acad Sci 1152:135–144
Kreft M et al (2003) Properties of exocytotic response in vertebrate photoreceptors. J Neurophysiol 90:218–225
Neher E, Marty A (1982) Discrete changes of cell membrane capacitance observed under conditions of enhanced secretion in bovine adrenal chromaffin cells. Proc Natl Acad Sci USA 79:6712–6716
Kreft M et al (2004) Properties of Ca(2+)-dependent exocytosis in cultured astrocytes. Glia 46:437–445
Bollmann JH et al (2000) Calcium sensitivity of glutamate release in a calyx-type terminal. Science 289:953–957
Heidelberger R et al (1994) Calcium dependence of the rate of exocytosis in a synaptic terminal. Nature 371:513–515
Schneggenburger R, Neher E (2000) Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature 406:889–893
Young SM, Neher E (2009) Synaptotagmin has an essential function in synaptic vesicle positioning for synchronous release in addition to its role as a calcium sensor. Neuron 63:482–496
Jean YY et al (2008) Glutamate elicits release of BDNF from basal forebrain astrocytes in a process dependent on metabotropic receptors and the PLC pathway. Neuron Glia Biol 4:35–42
Hua X et al (2004) Ca2+-dependent glutamate release involves two classes of endoplasmic reticulum Ca2+ stores in astrocytes. J Neurosci Res 76:86–97
MacVicar BA (1984) Voltage-dependent calcium channels in glial cells. Science 226:1345–1347
Yaguchi T, Nishizaki T (2010) Extracellular high K+ stimulates vesicular glutamate release from astrocytes by activating voltage-dependent calcium channels. J Cell Physiol 225:512–518
Akopian G et al (1996) Identified glial cells in the early postnatal mouse hippocampus display different types of Ca2+ currents. Glia 17:181–194
Golovina VA (2005) Visualization of localized store-operated calcium entry in mouse astrocytes. Close proximity to the endoplasmic reticulum. J Physiol 564:737–749
Malarkey EB et al (2008) Ca2+ entry through TRPC1 channels contributes to intracellular Ca2+ dynamics and consequent glutamate release from rat astrocytes. Glia 56:821–835
Shigetomi E et al (2011) TRPA1 channels regulate astrocyte resting calcium and inhibitory synapse efficacy through GAT-3. Nat Neurosci 15:70–80
Lalo U et al (2011) Ionotropic receptors in neuronal-astroglial signalling: what is the role of “excitable” molecules in non-excitable cells. Biochim Biophys Acta 1813:992–1002
Minelli A et al (2007) Cellular and subcellular localization of Na+-Ca2+ exchanger protein isoforms, NCX1, NCX2, and NCX3 in cerebral cortex and hippocampus of adult rat. Cell Calcium 41:221–234
Goldman WF et al (1994) Sodium/calcium exchange in rat cortical astrocytes. J Neurosci 14:5834–5843
Axelrod J (1974) Neurotransmitters. Sci Am 230:59–71
Do KQ et al (1997) Beta-Adrenergic stimulation promotes homocysteic acid release from astrocyte cultures: evidence for a role of astrocytes in the modulation of synaptic transmission. J Neurochem 68:2386–2394
Volterra A, Meldolesi J (2005) Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci 6:626–640
Martin ED et al (2007) Adenosine released by astrocytes contributes to hypoxia-induced modulation of synaptic transmission. Glia 55:36–45
Hertz L et al (1999) Astrocytes: glutamate producers for neurons. J Neurosci Res 57:417–428
Westergaard N et al (1996) Evaluation of the importance of transamination versus deamination in astrocytic metabolism of [U-13C]glutamate. Glia 17:160–168
Montana V et al (2004) Vesicular glutamate transporter-dependent glutamate release from astrocytes. J Neurosci 24:2633–2642
Fremeau RT Jr et al (2002) The identification of vesicular glutamate transporter 3 suggests novel modes of signaling by glutamate. Proc Natl Acad Sci USA 99:14488–14493
Anlauf E, Derouiche A (2005) Astrocytic exocytosis vesicles and glutamate: a high-resolution immunofluorescence study. Glia 49:96–106
Zhang Q et al (2004) Fusion-related release of glutamate from astrocytes. J Biol Chem 279:12724–12733
Bezzi P et al (2004) Astrocytes contain a vesicular compartment that is competent for regulated exocytosis of glutamate. Nat Neurosci 7:613–620
Crippa D et al (2006) Synaptobrevin2-expressing vesicles in rat astrocytes: insights into molecular characterization, dynamics and exocytosis. J Physiol 570:567–582
Wolosker H et al (1999) Purification of serine racemase: biosynthesis of the neuromodulator d-serine. Proc Natl Acad Sci USA 96:721–725
Rosenberg D et al (2010) Neuronal release of d-serine: a physiological pathway controlling extracellular d-serine concentration. FASEB J 24:2951–2961
Wolosker H (2011) Serine racemase and the serine shuttle between neurons and astrocytes. Biochim Biophys Acta 1814:1558–1566
Kartvelishvily E et al (2006) Neuron-derived d-serine release provides a novel means to activate N-methyl-d-aspartate receptors. J Biol Chem 281:14151–14162
Oliet SH, Mothet JP (2006) Molecular determinants of d-serine-mediated gliotransmission: from release to function. Glia 54:726–737
Henneberger C et al (2010) Long-term potentiation depends on release of d-serine from astrocytes. Nature 463:232–236
Vélez-Fort M et al (2011) Central role of GABA in neuron-glia interactions. Neuroscientist. doi:10.1177/1073858411403317
Angulo MC et al (2008) GABA, a forgotten gliotransmitter. Prog Neurobiol 86:297–303
Lee S et al (2010) Channel-mediated tonic GABA release from glia. Science 330:790–796
Echigo N, Moriyama Y (2004) Vesicular inhibitory amino acid transporter is expressed in gamma-aminobutyric acid (GABA)-containing astrocytes in rat pineal glands. Neurosci Lett 367:79–84
Unichenko P et al (2012) Intracellular Na+ concentration influences short-term plasticity of glutamate transporter-mediated currents in neocortical astrocytes. Glia 60:605–614
Araque A et al (1998) Calcium elevation in astrocytes causes an NMDA receptor-dependent increase in the frequency of miniature synaptic currents in cultured hippocampal neurons. J Neurosci 18:6822–6829
Liu T et al (2011) Calcium triggers exocytosis from two types of organelles in a single astrocyte. J Neurosci 31:10593–10601
Parpura V, Haydon PG (2000) Physiological astrocytic calcium levels stimulate glutamate release to modulate adjacent neurons. Proc Natl Acad Sci USA 97:8629–8634
Jahn R, Scheller R (2006) SNAREs-engines for membrane fusion. Nat Rev Mol Cell Biol 7:631–643
Stigliani S et al (2006) Glia re-sealed particles freshly prepared from adult rat brain are competent for exocytotic release of glutamate. J Neurochem 96:656–668
Montana V et al (2006) Vesicular transmitter release from astrocytes. Glia 54:700–715
Schubert V et al (2011) SNARE protein expression in synaptic terminals and astrocytes in the adult hippocampus: a comparative analysis. Glia 59:1472–1488
Chen X et al (2005) “Kiss-and-run” glutamate secretion in cultured and freshly isolated rat hippocampal astrocytes. J Neurosci 25:9236–9243
Zhang Q et al (2004) Synaptotagmin IV regulates glial glutamate release. Proc Natl Acad Sci USA 101:9441–9446
Maienschein V et al (1999) A plethora of presynaptic proteins associated with ATP-storing organelles in cultured astrocytes. Glia 26:233–244
Stenovec M et al (2007) Ca2+-dependent mobility of vesicles capturing anti-VGLUT1 antibodies. Exp Cell Res 313:3809–3818
Bergersen LH et al (2011) Immunogold detection of l-glutamate and d-serine in small synaptic-like microvesicles in adult hippocampal astrocytes. Cereb Cortex, Oxford
Xu J et al (2007) Glutamate-induced exocytosis of glutamate from astrocytes. J Biol Chem 282:24185–24197
Kang N et al (2005) Astrocytic glutamate release-induced transient depolarization and epileptiform discharges in hippocampal CA1 pyramidal neurons. J Neurophysiol 94:4121–4130
Bergersen LH, Gundersen V (2009) Morphological evidence for vesicular glutamate release from astrocytes. Neuroscience 158:260–265
Krzan M et al (2003) Calcium-dependent exocytosis of atrial natriuretic peptide from astrocytes. J Neurosci 23:1580–1583
Bowser DN, Khakh BS (2007) Two forms of single-vesicle astrocyte exocytosis imaged with total internal reflection fluorescence microscopy. Proc Natl Acad Sci USA 104:4212–4217
Domercq M et al (2006) P2Y1 receptor-evoked glutamate exocytosis from astrocytes: control by tumor necrosis factor-alpha and prostaglandins. J Biol Chem 281:30684–30696
Marchaland J et al (2008) Fast subplasma membrane Ca2+ transients control exo-endocytosis of synaptic-like microvesicles in astrocytes. J Neurosci 28:9122–9132
Del Castillo J, Katz B (1954) Quantal components of the end-plate potential. J Physiol 124:560–573
Pasti L et al (2001) Cytosolic calcium oscillations in astrocytes may regulate exocytotic release of glutamate. J Neurosci 21:477–484
Schell MJ et al (1995) d-serine, an endogenous synaptic modulator: localization to astrocytes and glutamate-stimulated release. Proc Natl Acad Sci USA 92:3948–3952
Mothet JP et al (2005) Glutamate receptor activation triggers a calcium-dependent and SNARE protein-dependent release of the gliotransmitter d-serine. Proc Natl Acad Sci USA 102:5606–5611
Martineau M et al (2008) Confocal imaging and tracking of the exocytotic routes for d-serine-mediated gliotransmission. Glia 56:1271–1284
Dannies PS (1999) Protein hormone storage in secretory granules: mechanisms for concentration and sorting. Endocr Rev 20:3–21
Calegari F et al (1999) A regulated secretory pathway in cultured hippocampal astrocytes. J Biol Chem 274:22539–22547
Ramamoorthy P, Whim MD (2008) Trafficking and fusion of neuropeptide y-containing dense-core granules in astrocytes. J Neurosci 28:13815–13827
McKenzie JC et al (2001) Atrial natriuretic peptide-like immunoreactivity in neurons and astrocytes of human cerebellum and inferior olivary complex. J Histochem Cytochem 49:1453–1467
Potter LR et al (2006) Natriuretic peptides, their receptors, and cyclic guanosine monophosphate-dependent signaling functions. Endocr Rev 27:47–72
Skowrońska M et al (2010) Stimulation of natriuretic peptide receptor C attenuates accumulation of reactive oxygen species and nitric oxide synthesis in ammonia-treated astrocytes. J Neurochem 115:1068–1076
Pangrsic T et al (2007) Exocytotic release of ATP from cultured astrocytes. J Biol Chem 282:28749–28758
Coco S et al (2003) Storage and release of ATP from astrocytes in culture. J Biol Chem 278:1354–1362
Stenovec M et al (2004) Slow spontaneous secretion from single large dense-core vesicles monitored in neuroendocrine cells. FASEB J 18:1270–1272
Baertschi AJ et al (2001) Acid prohormone sequence determines size, shape, and docking of secretory vesicles in atrial myocytes. Circ Res 89:E23–E29
Potokar M et al (2008) Stimulation inhibits the mobility of recycling peptidergic vesicles in astrocytes. Glia 56:135–144
Potokar M et al (2007) Cytoskeleton and vesicle mobility in astrocytes. Traffic 8:12–20
Potokar M et al (2005) Vesicle mobility studied in cultured astrocytes. Biochem Biophys Res Commun 329:678–683
Potokar M et al (2010) Intermediate filaments attenuate stimulation-dependent mobility of endosomes/lysosomes in astrocytes. Glia 58:1208–1219
Potokar M et al (2011) Physiopathologic dynamics of vesicle traffic in astrocytes. Histol Histopathol 26:277–284
Stenovec M et al (2011) Amyotrophic lateral sclerosis immunoglobulins G enhance the mobility of Lysotracker-labelled vesicles in cultured rat astrocytes. Acta Physiol (Oxf) 203:457–471
Kopan R, Ilagan MX (2009) The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137:216–233
Soos JM et al (1998) Astrocytes express elements of the class II endocytic pathway and process central nervous system autoantigen for presentation to encephalitogenic T cells. J Immunol 161:5959–5966
Robinson MB (2002) Regulated trafficking of neurotransmitter transporters: common notes but different melodies. J Neurochem 80:1–11
Stenovec M et al (2008) EAAT2 density at the astrocyte plasma membrane and Ca(2+)-regulated exocytosis. Mol Membr Biol 25:203–215
Bergami M et al (2008) Uptake and recycling of pro-BDNF for transmitter-induced secretion by cortical astrocytes. J Cell Biol 183:213–221
Fields RD, Burnstock G (2006) Purinergic signalling in neuron-glia interactions. Nat Rev Neurosci 7:423–436
Verkhratsky A et al (2009) Purinoceptors on neuroglia. Mol Neurobiol 39:190–208
Prado J et al (2010) Glial cells as sources and targets of natriuretic peptides. Neurochem Int 57:367–374
Sawada K et al (2008) Identification of a vesicular nucleotide transporter. Proc Natl Acad Sci USA 105:5683–5686
Larsson M et al (2011) Functional and Anatomical Identification of a vesicular transporter mediating neuronal ATP release. Cereb Cortex, Oxford
Bal-Price A et al (2002) Nitric oxide induces rapid, calcium-dependent release of vesicular glutamate and ATP from cultured rat astrocytes. Glia 40:312–323
Abdipranoto A et al (2003) Mechanisms of secretion of ATP from cortical astrocytes triggered by uridine triphosphate. NeuroReport 14:2177–2181
Pryazhnikov E, Khiroug L (2008) Sub-micromolar increase in [Ca2+]i triggers delayed exocytosis of ATP in cultured astrocytes. Glia 56:38–49
Halassa MM et al (2009) Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron 61:213–219
Zhang Z et al (2007) Regulated ATP release from astrocytes through lysosome exocytosis. Nat Cell Biol 9:945–953
Verderio C et al (2011) TI-VAMP/VAMP7 is the snare of secretory lysosomes contributing to ATP secretion from astrocytes. Biol Cell 104:213–228
Potokar M et al (2012) Rab4 and Rab5 GTPase are required for directional mobility of endocytic vesicles in astrocytes. Glia 60:594–604
Fontana A et al (1984) Astrocytes present myelin basic protein to encephalitogenic T-cell lines. Nature 307:273–276
Pekny M, Pekna M (2004) Astrocyte intermediate filaments in CNS pathologies and regeneration. J Pathol 204:428–437
Junyent F et al (2011) Content and traffic of taurine in hippocampal reactive astrocytes. Hippocampus 21:185–197
Zhou J, Sutherland ML (2004) Glutamate transporter cluster formation in astrocytic processes regulates glutamate uptake activity. J Neurosci 24:6301–6306
Nakagawa T et al (2008) Mechanisms of substrate transport-induced clustering of a glial glutamate transporter GLT-1 in astroglial-neuronal cultures. Eur J Neurosci 28:1719–1730
Leterrier C et al (2004) Constitutive endocytic cycle of the CB1 cannabinoid receptor. J Biol Chem 279:36013–36021
Navarrete M, Araque A (2008) Endocannabinoids mediate neuron-astrocyte communication. Neuron 57:883–893
Bezzi P et al (2001) CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat Neurosci 4:702–710
Osborne KD et al (2009) Dynamic imaging of cannabinoid receptor 1 vesicular trafficking in cultured astrocytes. ASN Neuro 1(5):art:e00022. doi:10.1042/AN20090040
Calì C et al (2008) SDF 1-alpha (CXCL12) triggers glutamate exocytosis from astrocytes on a millisecond time scale: imaging analysis at the single-vesicle level with TIRF microscopy. J Neuroimmunol 198:82–91
Calì C, Bezzi P (2010) CXCR4-mediated glutamate exocytosis from astrocytes. J Neuroimmunol 224:13–21
Stellwagen D et al (2005) Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci 25:3219–3228
Parpura V et al (2010) Regulated exocytosis in astrocytic signal integration. Neurochem Int 57:451–459
Pascual O (2005) Astrocytic purinergic signaling coordinates synaptic networks. Science 310:113–116
Cali C et al (2008) SDF 1-alpha (CXCL12) triggers glutamate exocytosis from astrocytes on a millisecond time scale: imaging analysis at the single-vesicle level with TIRF microscopy. J Neuroimmunol 198:82–91
Malarkey EB, Parpura V (2011) Temporal characteristics of vesicular fusion in astrocytes: examination of synaptobrevin 2-laden vesicles at single vesicle resolution. J Physiol 589:4271–4300
Jorgacevski J et al (2010) Fusion pore stability of peptidergic vesicles. Mol Membr Biol 27:65–80
Jorgacevski J et al (2011) Munc18-1 tuning of vesicle merger and fusion pore properties. J Neurosci 31:9055–9066
Vardjan N et al (2007) Subnanometer fusion pores in spontaneous exocytosis of peptidergic vesicles. J Neurosci 27:4737–4746
Acknowledgments
This work was supported by the grants P3 310, J3 4051, J3 3632 and J3 4146 from the Slovenian Research Agency (ARRS), CipKeBip and the EduGlia ITN EU grant.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Special issue: In honor of Leif Hertz.
Alenka Guček, Nina Vardjan contributed equally to this work.
Rights and permissions
About this article
Cite this article
Guček, A., Vardjan, N. & Zorec, R. Exocytosis in Astrocytes: Transmitter Release and Membrane Signal Regulation. Neurochem Res 37, 2351–2363 (2012). https://doi.org/10.1007/s11064-012-0773-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-012-0773-6