
Abstract
Pro-inflammatory and anti-inflammatory mediators derived from arachidonic acid (AA) modulate peripheral inflammation and its resolution. Aspirin (ASA) is a unique non-steroidal anti-inflammatory drug, which switches AA metabolism from prostaglandin E2 (PGE2) and thromboxane B2 (TXB2) to lipoxin A4 (LXA4) and 15-epi-LXA4. However, it is unknown whether chronic therapeutic doses of ASA are anti-inflammatory in the brain. We hypothesized that ASA would dampen increases in brain concentrations of AA metabolites in a rat model of neuroinflammation, produced by a 6-day intracerebroventricular infusion of bacterial lipopolysaccharide (LPS). In rats infused with LPS (0.5 ng/h) and given ASA-free water to drink, concentrations in high-energy microwaved brain of PGE2, TXB2 and leukotriene B4 (LTB4) were elevated. In rats infused with artificial cerebrospinal fluid, 6 weeks of treatment with a low (10 mg/kg/day) or high (100 mg/kg/day) ASA dose in drinking water decreased brain PGE2, but increased LTB4, LXA4 and 15-epi-LXA4 concentrations. Both doses attenuated the LPS effects on PGE2, and TXB2. The increments in LXA4 and 15-epi-LXA4 caused by high-dose ASA were significantly greater in LPS-infused rats. The ability of ASA to increase anti-inflammatory LXA4 and 15-epi-LXA4 and reduce pro-inflammatory PGE2 and TXB2 suggests considering aspirin further for treating clinical neuroinflammation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
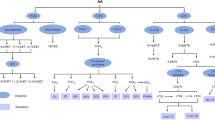
Aspirin [acetylsalicylic acid, ASA], a non-steroidal anti-inflammatory drug (NSAID), is used widely to relieve pain, fever and peripheral inflammation. Low-dose ASA (75–150 mg/day) is recommended for long-term prophylaxis of thrombotic events such as heart attacks and strokes, while a higher dose (1 g) has analgesic and antipyretic effects [1]. ASA irreversibly inhibits cyclooxygenase (COX)-1, which converts arachidonic acid (AA, 20:4n-6) to prostaglandin endoperoxides, and thus reduces prostaglandin (PG) and thromboxane (TX) formation [2] (Fig. 1). ASA also acetylates COX-2 [3], which converts AA to 15(R)-hydroxyeicosatetraenoic acid (HETE), which then can be metabolized by 5-lipoxygenase (5-LOX) to 15-epimeric lipoxin (LX) A4 and B4 (15-epi-LX) in leukocytes and endothelial cells [4]. Lipoxins, generated by the actions of 5- and 12-LOX or of 15- and 5-LOX, and 15-epi-LX play key roles in resolution of the inflammatory reaction [5–8]. Other NSAIDs are unable to generate 15-epi-LXA4, and selective COX-2 inhibitors like celecoxib prevent ASA-induced 15-epi-LXA4 [4, 9, 10].

Main pathways of eicosanoid biosynthesis and ASA effects. AA is converted into PGH2 via COX, and subsequently into PGE2 and TXB2 via PGE synthase and TX synthase, respectively. ASA inhibits COX-1 and acetylates COX-2. Acetylated COX-2 converts AA into 15(R)-HETE, which is metabolized further by 5-LOX into 15-epi-LXA4. 5-LOX converts AA into 5(S)-HpETE, and subsequently, into LTA4, which is converted into LTB4 by LTA4 hydrolase. Alternatively, in the presence of 12-LOX, LTA4 might be converted into LXA4. Another pathway to LX biosynthesis involves the conversion of AA by 15-LOX into 15(S)-HETE, which is transformed by 5-LOX into LXA4 [2, 5, 7, 18]. AA arachidonic acid, COX cyclooxygenase, HETE hydroxyeicosatetraenoic acid, HpETE hydroperoxyeicosatetraenoic acid, LOX lipoxygenase, LT leukotriene, LX lipoxin, PG prostaglandin, TX thromboxane
Neuroinflammation is reported to contribute to a number of human psychiatric, neurodegenerative, viral and ischemic brain diseases, including Alzheimer’s disease, bipolar disorder, stroke, and HIV-1 dementia [11–15]. In rats, neuroinflammation can be produced by chronic intracerebroventricular (icv) infusion of bacterial lipopolysaccharide (LPS) at a rate of 1 ng/h [16], or at a higher rate of 250 ng/h [17]. We reported that a 6-day icv infusion of low-dose LPS (0.5 ng/h) in rats increased markers of the AA metabolic cascade [18] in brain: activities of AA-selective Ca2+-dependent cytosolic phospholipase A2 (cPLA2) and of secretory sPLA2, AA turnover in brain phospholipids, and brain concentrations of unesterified AA and of its PGE2 and TXB2 metabolites. Net brain COX activity and COX-1 and COX-2 protein levels were not changed significantly [19–22]. Many of the changes caused by LPS were prevented by 6-week LiCl feeding [19]. The same low-dose LPS infused for 6 days increased lectin-reactive microglia, changed the morphology of glial fibrillary acidic protein-positive astrocytes [21], and increased protein levels of tumor necrosis factor-alpha (TNF-α) and inducible nitric oxide synthase without altering interleukin (IL)-1β protein (Kellom M. and Rapoport S.I., unpublished observations). TNF-α has been shown to regulate cPLA2 sPLA2 and COX-2 expression [23–25]. Thus, in this LPS model, altered brain AA metabolism is a major participant in the neuroinflammatory process.
Despite reported ASA effects on peripheral inflammation, it is unknown whether chronic therapeutic doses of ASA are anti-inflammatory in the brain. We therefore thought it of interest in this paper to examine the effects of chronic ASA on brain AA eicosanoids in the LPS neuroinflammation model, when using high-energy microwaving to prevent postmortem release and metabolism of fatty acids [26]. Because brain concentrations of PGE2 and TXB2, derived from AA via COX-1 and COX-2, were increased significantly by 6-day low-dose LPS infusion, we hypothesized that chronic ASA would dampen these increments and perhaps trigger formation of anti-inflammatory mediators. We quantified PGE2, TXB2, LTB4, LXA4 and 15-epi-LXA4 concentrations by ELISA in high-energy microwaved brain from rats given for 6 weeks a low-dose (10 mg/kg/day) or high-dose (100 mg/kg/day) of ASA in their drinking water or ASA-free water, and infused icv LPS at a rate of 0.5 ng/h, or artificial cerebrospinal fluid (aCSF), for 6 days [19–22]. An interspecies dose conversion factor based on body surface (7:1 for conversion from rat to human) [27] indicated that the two ASA doses were equivalent to daily human doses of 1.43 mg/kg and 14.3 mg/kg respectively, or 100 mg and 1 g respectively, for a 70 kg person.
Experimental Procedure
Animals
Experiments were performed under a protocol approved by the Animal Care and Use Committee of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, in accordance with National Institutes of Health Guidelines on the Care and Use of Laboratory Animals (NIH Publication No. 86-23). Two-month-old male Fischer 344 rats (n = 48) (Taconic Farms, Rockville, MD) were acclimated for 1 week in an animal facility in which temperature, humidity and light–dark cycle were regulated. The rats had ad libitum access to water and food (Rodent NIH-31 auto 18-4 diet, Zeigler Bros, Gardners, PA). The diet contained (as % of total fatty acid) 20.1% saturated, 22.5% monounsaturated, 47.9% linoleic, 5.1% α-linolenic, 0.02% AA, 2.0% eicosapentaenoic, and 2.3% docosahexaenoic acid [28]. The animals had free access to drinking water, and rats treated with low-dose ASA (n = 16; 10 mg/kg/day) and high-dose ASA (n = 16; 100 mg/kg/day) received the drug in their drinking water for 42 days. Water consumption and body weight were measured twice a week to calculate daily ASA intake and to adjust the ASA concentration in the drinking water.
Surgery
After receiving plain water or water containing ASA, a rat was anesthetized and an indwelling cerebroventricular cannula was fixed in place as described [19–22]. aCSF or low-dose LPS (1 μg/ml dissolved in aCSF, 0.5 ng/h; Sigma, Saint Louis, MO; Escherichia coli, serotype 055:B5) was infused into the fourth ventricle through the cannula connected to an osmotic pump (Alzet®, Model 2002, Cupertino, CA). Before surgery, the prefilled pump was placed in sterile 0.9% NaCl at 37°C overnight to start immediate pumping. Postoperative care included triple antibiotic ointment (Perrigo, Allegan, MI) applied to the wound, and 5 ml of sterile isotonic saline (s.c., 0.9% NaCl) to prevent dehydration during recovery from anesthesia. Following 6-day LPS or aCSF icv infusion, the rat was anesthetized with Nembutal® (40 mg/kg i.p.) and subjected to head-focused high-energy microwave irradiation (5.5 kW, 3.4 s; Cober Electronics, Stamford, CT) to denature brain enzymes and stop metabolism [26, 29]. The head was cooled in dry ice, the brain was excised, frozen in 2-methylbutane maintained at −40°C, and stored at −80°C until analyzed.
Brain Eicosanoid Measurement by ELISA
Microwaved brains were prepared and analyzed for eicosanoid measurements as previously described [19] using PGE2, TXB2, LTB4, LXA4 and 15-epi-LXA4 ELISA assay kits (Oxford Biochemical Research, Oxford, MI).
Statistical Analysis
A two-way analysis of variance (ANOVA), comparing ASA administration (ASA vs. ASA-free water) with LPS infusion (LPS vs. aCSF) was performed using SPSS 16.0 X (SPSS Inc., Chicago, IL). If ASA × LPS interactions were statistically insignificant, probabilities of main effects of ASA and LPS were reported. If interactions were statistically significant, these probabilities were not reported because they cannot be interpreted with surety [30]. A one-way ANOVA with Bonferroni’s post-hoc test was performed with correction for six comparisons (effect of LPS or aCSF in ASA-free water and ASA-treated rats, and ASA 100 mg/kg/day vs. 10 mg/kg/day in aCSF-infused rats). Data are reported as mean ± SD (n = 8), with statistical significance set at P ≤ 0.05.
Results
Dose Calculations, Water Consumption and Body Weight
During the period of 6 weeks, all rats consumed 19–20 ml/day of water (data not shown). The calculated net weekly ASA intake equaled 9–11 mg/kg/day (low-dose) or 95–110 mg/kg/day (high-dose). Chronic ASA administration [31, 32] and 6-day LPS infusion [19, 22] were well tolerated by all rats for the duration of the study. Neither ASA nor 6-day LPS infusion significantly influenced body weight or water consumption (data not shown).
Brain Eicosanoids
Prostaglandin E2
A two-way ANOVA showed a statistically significant ASA × LPS interaction (P < 0.001) with regard to brain PGE2 concentration. Subsequent one-way ANOVA with Bonferroni post-hoc tests showed that LPS compared with aCSF infusion significantly increased PGE2 by 60% (P < 0.001) in rats given ASA-free water. ASA at 10 and 100 mg/kg/day decreased basal brain PGE2 by 46% (P < 0.01) and 85% (P < 0.001) respectively in aCSF-infused rats, and prevented the significant increase in PGE2 in LPS-infused rats (P > 0.05) (Fig. 2a). High-dose ASA significantly decreased PGE2 more than did low-dose ASA in aCSF-infused rats (P < 0.05).

Effects of LPS and ASA on brain concentrations of: a PGE2; b TXB2; c LTB4; d LXA4 and e 15-epi-LXA4. Data are means ± SD (n = 8) and were analyzed with a two-way ANOVA. * P < 0.05, ** P < 0.01, *** P < 0.001 versus rats given ASA-free water and infused with aCSF; # P < 0.05, ## P < 0.01 ASP 100 mg/kg/day versus ASP 10 mg/kg/day in aCSF-infused rats; ∆ P < 0.05, ASP 100 mg/kg/day + LPS vs. ASP 100 mg/kg/day + aCSF
Thromboxane B2
A two-way ANOVA showed a significant ASA × LPS interaction (P = 0.001). Subsequent one-way ANOVA with Bonferroni post-hoc tests showed that LPS compared with aCSF infusion significantly increased brain TXB2 by 2.5-fold (P < 0.001) in rats given ASA-free water. ASA alone (10 or 100 mg/kg/day) had no effect on TXB2 (P > 0.05) in aCSF-infused rats. However, both ASA doses significantly blocked the LPS-induced TXB2 increment (Fig 2b).
Leukotriene B4
A two-way ANOVA showed significant ASA (P < 0.001) and LPS (P = 0.03) effects without a significant ASA × LPS interaction (P = 0.23), indicating that ASA did not alter the LPS response. A Bonferroni post-hoc test indicated that LPS infusion, and ASA at 100 mg/kg/day increased LTB4 concentration by 10- and 19-fold (P < 0.01), respectively compared to aCSF-infused rats given ASA-free water, whereas ASA at 10 mg/kg/day had no significant effect. ASA did not prevent the significant LPS-induced LTB4 increase (Fig 2c).
Lipoxin A4
A two-way ANOVA showed a significant ASA × LPS interaction (P < 0.001) for LXA4. Subsequent one-way ANOVA with Bonferroni post-hoc tests showed that LPS compared with aCSF infusion had no effect on LXA4 (P > 0.05) in rats given ASA-free water. ASA at 10 and 100 mg/kg/day increased LXA4 by twofold in aCSF-infused rats. ASA at 100 mg/kg/day augmented further LXA4 production by 25% in LPS-infused rats (P < 0.05) (Fig. 2d).
15-Epi-Lipoxin A4
A two-way ANOVA showed significant ASA (P < 0.001) and LPS (P < 0.04) effects without a significant ASA × LPS interaction (P = 0.10) for 15-epi-LXA4. Bonferroni post-hoc tests indicated that in rats given ASA-free water, LPS compared with aCSF infusion had no effect (P > 0.05). ASA at 10 mg/kg/day and at 100 mg/kg/day increased the 15-epi-LXA4 concentration by 2.6-fold (P < 0.05) and 3.7-fold (P < 0.001), respectively, in aCSF-infused rats. ASA 100 mg/kg/day augmented 15-epi-LXA4 by 50% (P < 0.01) in LPS-infused rats (Fig 2e).
Discussion
Six days of icv LPS infusion in adult rats at a rate of 0.5 ng/h, compared with aCSF infusion, significantly increased brain concentrations of PGE2, TXB2 and LTB4. Six weeks of low (10 mg/kg/day) and/or high (100 mg/kg/day) doses of ASA in water compared with ASA-free drinking water significantly decreased the brain PGE2 concentration while increasing basal LTB4, LXA4 and 15-epi-LXA4 concentrations in aCSF-infused rats. Both chronic ASA doses attenuated the LPS-induced increments in PGE2 and TXB2. The increments in LXA4 and 15-epi-LXA4 in rats consuming high-dose ASA were significantly greater in LPS-infused than aCSF-infused rats.
The ability of both doses of ASA to prevent the elevations in brain PGE2 and TXB2 caused by LPS infusion in rats drinking ASA-free water, suggests that ASA enters brain and inhibits COX activity when given at the clinically relevant doses [2]. Similarly, chronic ASA reduced brain PGE2 elevations in the experimental mouse antiphospholipid syndrome, which is associated with neuroinflammation [33]. Although chronic ASA inhibited brain PGE2 formation in aCSF-infused rats, it did not change significantly the basal brain TXB2 concentration. The brain PGE2 concentration was 1,900-fold greater than the TXB2 concentration in the rats infused with aCSF and consuming ASA-free drinking water. This finding suggests that COX-2 is the predominant isoform in the normal unstimulated rat brain [34].
The brain concentration of pro-inflammatory LTB4, the AA product of 5-LOX and LTA4 hydrolase [35], was increased by LPS but also by high-dose ASA, suggesting that both exposures activated the 5-LOX pathway (Fig. 1). Consistent with this observation, an increased LTB4 concentration following ASA has been reported in the mouse hippocampus [36] and in other rodent tissues [37–39], as well as in humans [40]. LTB4 is known to activate cerebellar ryanodine receptors, which can mobilize Ca2+ from the endoplasmic reticulum [41], target activated leukocytes at a site of neuroinflammation, and induce adhesion molecules on endothelial cells and neutrophils [35, 38]. Furthermore, LTB4 stimulated proliferation and differentiation of neural stem cells into neurons [42], which might be relevant in pathophysiological disorders with neuroinflammation.
In this study, the brain was subjected to high-energy microwave irradiation to rapidly and irreversibly inactivate brain enzymes, thereby avoiding ischemic release of unesterified fatty acids and other metabolic processes [26, 29, 43]. In the rats that consumed ASA-free water and infused with aCSF, the measured brain concentrations of PGE2 and TXB2 are consistent with reported values obtained with ELISA on similarly microwaved brain [19], although much lower than in non-microwaved brain [43, 44]. The control brain LTB4 concentration (0.11 ng/g) also agrees with reported values in microwaved brain [45, 46].
Both low- and high-dose ASA triggered formation of brain LXA4 and 15-epi-LXA4 in rats infused with aCSF, and rats treated with high-dose ASA and infused with LPS had higher brain concentrations of these two anti-inflammatory mediators, suggesting some redirection of AA metabolism from PGE2 and TXB2 to LXA4 and 15-epi-LXA4 biosynthesis [4]. The increased 15-epi-LXA4 concentrations suggest COX-2 acetylation by ASA and increased 5-LOX activity in rat brain. In comparison, low-dose ASA increased 15-epi-LXA4 in plasma of healthy volunteers [47], and in response to acute inflammation in human blood leukocytes [48]. Finding LXA4 in control rat brain is consistent with other reports [36, 49, 50].
There are few reports concerning LXA4 or 15-epi-LXA4 functions in brain [6, 7]. LXA4 inhibited IL-8 and ICAM-1 expression induced by IL-1β through a NF-κΒ dependent mechanism in human astrocytoma cells [51]. In macrophages, LXA4 reduced LPS-induced TNF-α by inhibiting activation of NF-κΒ, which is a transcriptional factor for the cPLA2, sPLA2 and COX-2 genes [52–55]. LXA4 also was neuroprotective by acting as an agonist of peroxisome proliferator-activated receptor-γ in a rat stroke model [50].
Although both ASA doses altered brain eicosanoid concentrations in aCSF-infused rats, a dose-dependent response was not observed except for the PGE2 concentration, which may be explained by the nonlinear pharmacokinetics of ASA in rats [56]. Because both ASA doses normalized PGE2 and TXB2 in LPS-infused rats, and only the high-dose ASA increased the increments in LXA4 and 15-epi-LXA4, different doses of ASA may be necessary to inhibit the constitutive COX-1 and to acetylate the inducible COX-2.
In summary, we have shown that chronic ASA, when given in drinking water to rats at a clinically relevant low- or high-dose, enters brain and triggers formation of AA-derived anti-inflammatory mediators, LXA4 or 15-epi-LXA4. Additionally, chronic ASA has a significant impact on eicosanoids associated with LPS-induced neuroinflammation. Similarly, 30-day nitro-ASA administration attenuated neuroinflammation in rats infused chronically with LPS [57]. ASA also has been reported to be neuroprotective against cerebral ischemia [58], and to improve spatial learning in rats [32]. Chronic low-dose ASA (5 mg/kg/day) provided cerebrovascular protection from oxidant damage in rats [59]. In humans, chronic low-dose ASA was beneficial in Parkinson’s disease [60], ameliorated mood [61], and reduced morbidity in (presumably) bipolar disorder patients on lithium [62]. Adjuvant ASA therapy also reduced symptoms of schizophrenia spectrum disorders in a randomized, double-blind, placebo-controlled trial [63]. In view of the multiple reported central effects of low- and high-dose ASA, trials with ASA might be further considered for brain diseases associated with neuroinflammation [11–14, 62–64]. Future studies of the effects of long-term LPS infusion with ASA, and a detailed analysis of brain markers of neuroinflammation would be informative and clinically relevant.
Abbreviations
- AA:
-
Arachidonic acid
- aCSF:
-
Artificial cerebrospinal fluid
- COX:
-
Cyclooxygenase
- ELISA:
-
Enzyme-linked immunosorbent assay
- HETE:
-
Hydroxyeicosatetraenoic acid
- icv:
-
Intracerebroventricular
- IL:
-
Interleukin
- LPS:
-
Lipopolysaccharide
- LT:
-
Leukotriene
- LXA4 :
-
Lipoxin A4
- 15-epi-LXA4 :
-
15-epimeric-lipoxin A4
- LOX:
-
Lipoxygenase
- PLA2 :
-
Phospholipase A2
- cPLA2 :
-
Cytosolic PLA2
- sPLA2 :
-
Secretory PLA2
- PGE2 :
-
Prostaglandin E2
- TXB2 :
-
Thromboxane B2
- NSAID:
-
Non-steroidal anti-inflammatory drug
References
Weissmann G (1991) Aspirin. Sci Am 264:84–90
Vane JR (1971) Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol 231:232–235
Meade EA, Smith WL, DeWitt DL (1993) Differential inhibition of prostaglandin endoperoxide synthase (cyclooxygenase) isozymes by aspirin and other non-steroidal anti-inflammatory drugs. J Biol Chem 268:6610–6614
Clària J, Serhan CN (1995) Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc Natl Acad Sci USA 92:9475–9479
Romano M, Chen XS, Takahashi Y et al (1993) Lipoxin synthase activity of human platelet 12-lipoxygenase. Biochem J 296(Pt 1):127–133
Chiang N, Arita M, Serhan CN (2005) Anti-inflammatory circuitry: lipoxin, aspirin-triggered lipoxins and their receptor ALX. Prostaglandins Leukot Essent Fatty Acids 73:163–177
Serhan CN (1997) Lipoxins and novel aspirin-triggered 15-epi-lipoxins (ATL): a jungle of cell-cell interactions or a therapeutic opportunity? Prostaglandins 53:107–137
Serhan CN (2005) Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot Essent Fatty Acids 73:141–162
Fiorucci S, de Lima Jr OM, Mencarelli A et al (2002) Cyclooxygenase-2-derived lipoxin A4 increases gastric resistance to aspirin-induced damage. Gastroenterology 123:1598–1606
Titos E, Chiang N, Serhan CN et al (1999) Hepatocytes are a rich source of novel aspirin-triggered 15-epi-lipoxin A4. Am J Physiol 277:C870–877
Dirnagl U, Iadecola C, Moskowitz MA (1999) Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci 22:391–397
Esposito G, Giovacchini G, Liow JS et al (2008) Imaging neuroinflammation in Alzheimer’s disease with radiolabeled arachidonic acid and PET. J Nucl Med 49:1414–1421
Minghetti L (2005) Role of inflammation in neurodegenerative diseases. Curr Opin Neurol 18:315–321
Rao JS, Harry GJ, Rapoport SI et al (2010) Increased excitotoxicity and neuroinflammatory markers in postmortem frontal cortex from bipolar disorder patients. Mol Psychiatry 15:384–392
Basselin M, Ramadan E, Igarashi M et al. (in press) Imaging upregulated brain arachidonic acid metabolism in HIV-1 transgenic rats. J Cereb Blood Flow Metab. Online (28 July 2010), doi:10.1038/jcbfm.2010.111
Richardson RL, Kim EM, Gardiner T et al (2005) Chronic intracerebroventricular infusion of lipopolysaccharide: effects of ibuprofen treatment and behavioural and histopathological correlates. Behav Pharmacol 16:531–541
Hauss-Wegrzyniak B, Dobrzanski P, Stoehr JD et al (1998) Chronic neuroinflammation in rats reproduces components of the neurobiology of Alzheimer’s disease. Brain Res 780:294–303
Shimizu T, Wolfe LS (1990) Arachidonic acid cascade and signal transduction. J Neurochem 55:1–15
Basselin M, Villacreses NE, Lee HJ et al (2007) Chronic lithium administration attenuates up-regulated brain arachidonic acid metabolism in a rat model of neuroinflammation. J Neurochem 102:761–772
Lee H, Villacreses NE, Rapoport SI et al (2004) In vivo imaging detects a transient increase in brain arachidonic acid metabolism: a potential marker of neuroinflammation. J Neurochem 91:936–945
Rosenberger TA, Villacreses NE, Hovda JT et al (2004) Rat brain arachidonic acid metabolism is increased by a 6-day intracerebral ventricular infusion of bacterial lipopolysaccharide. J Neurochem 88:1168–1178 Erratum in: J Neurochem (2004) 1190, 1255
Basselin M, Kim HW, Chen M et al (2010) Lithium modifies brain arachidonic and docosahexaenoic metabolism in rat lipopolysaccharide model of neuroinflammation. J Lipid Res 51:1049–1056
Bauer MK, Lieb K, Schulze-Osthoff K et al (1997) Expression and regulation of cyclooxygenase-2 in rat microglia. Eur J Biochem 243:726–731
Hoeck WG, Ramesha CS, Chang DJ et al (1993) Cytoplasmic phospholipase A2 activity and gene expression are stimulated by tumor necrosis factor: dexamethasone blocks the induced synthesis. Proc Natl Acad Sci USA 90:4475–4479
Arbibe L, Vial D, Rosinski-Chupin I et al (1997) Endotoxin induces expression of type II phospholipase A2 in macrophages during acute lung injury in guinea pigs: involvement of TNFα in lipopolysaccharide-induced type II phospholipase A2 synthesis. J Immunol 159:391–400
Deutsch J, Rapoport SI, Purdon AD (1997) Relation between free fatty acid and acyl-CoA concentrations in rat brain following decapitation. Neurochem Res 22:759–765
Voisin EM, Ruthsatz M, Collins JM et al (1990) Extrapolation of animal toxicity to humans: interspecies comparisons in drug development. Regul Toxicol Pharmacol 12:107–116
DeMar JC Jr, Lee HJ, Ma K et al (2006) Brain elongation of linoleic acid is a negligible source of the arachidonate in brain phospholipids of adult rats. Biochim Biophys Acta 1761:1050–1059
Farias SE, Basselin M, Chang L et al (2008) Formation of eicosanoids, E2/D2-isoprostanes and docosanoids following decapitation-induced ischemia, measured in high-energy microwaved rat brain. J Lipid Res 49:1990–2000
Tabachnick BG, Fidell LS (2001) Computer-assisted research design and analysis. Allyn and Bacon, Boston, pp 184–188
Renna NF, Vazquez MA, Lama MC et al (2009) Effect of chronic aspirin administration on an experimental model of metabolic syndrome. Clin Exp Pharmacol Physiol 36:162–168
Smith JW, Al-Khamees O, Costall B et al (2002) Chronic aspirin ingestion improves spatial learning in adult and aged rats. Pharmacol Biochem Behav 71:233–238
Tanne D, Katzav A, Beilin O et al (2008) Interaction of inflammation, thrombosis, aspirin and enoxaparin in CNS experimental antiphospholipid syndrome. Neurobiol Dis 30:56–64
Yamagata K, Andreasson KI, Kaufmann WE et al (1993) Expression of a mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoids. Neuron 11:371–386
Samuelsson B, Dahlen SE, Lindgren JA et al (1987) Leukotrienes and lipoxins: structures, biosynthesis, and biological effects. Science 237:1171–1176
Marcheselli VL, Hong S, Lukiw W et al (2003) Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J Biol Chem 278:43807–43817
Xiao L, Patterson PS, Yang C et al (1999) Role of eicosanoids in the pathogenesis of murine cerebral malaria. Am J Trop Med Hyg 60:668–673
Fiorucci S, Distrutti E, Mencarelli A et al (2003) Evidence that 5-lipoxygenase and acetylated cyclooxygenase 2-derived eicosanoids regulate leukocyte-endothelial adherence in response to aspirin. Br J Pharmacol 139:1351–1359
Planagumà A, Titos E, López-Parra M et al (2002) Aspirin (ASA) regulates 5-lipoxygenase activity and peroxisome proliferator-activated receptor alpha-mediated CINC-1 release in rat liver cells: novel actions of lipoxin A4 (LXA4) and ASA-triggered 15-epi-LXA4. FASEB J. 16:1937–1939
Engstrom K, Wallin R, Saldeen T (2001) Effect of low-dose aspirin in combination with stable fish oil on whole blood production of eicosanoids. Prostaglandins Leukot Essent Fatty Acids 64:291–297
Striggow F, Ehrlich BE (1997) Regulation of intracellular calcium release channel function by arachidonic acid and leukotriene B4. Biochem Biophys Res Commun 237:413–418
Wada K, Arita M, Nakajima A et al (2006) Leukotriene B4 and lipoxin A4 are regulatory signals for neural stem cell proliferation and differentiation. FASEB J 20:1785–1792
Golovko MY, Murphy EJ (2008) An improved LC-MS/MS procedure for brain prostanoid analysis using brain fixation with head-focused microwave irradiation and liquid-liquid extraction. J Lipid Res 49:893–902
Bosisio E, Galli C, Galli G et al (1976) Correlation between release of free arachidonic acid and prostaglandin formation in brain cortex and cerebellum. Prostaglandins 11:773–781
Ghelardoni S, Tomita YA, Bell JM et al (2004) Chronic carbamazepine selectively downregulates cytosolic phospholipase A2 expression and cyclooxygenase activity in rat brain. Biol. Psychiatry 56:248–254
Bosetti F, Weerasinghe GR, Rosenberger TA et al (2003) Valproic acid down-regulates the conversion of arachidonic acid to eicosanoids via cyclooxygenase-1 and -2 in rat brain. J Neurochem 85:690–696
Chiang N, Bermudez EA, Ridker PM et al (2004) Aspirin triggers antiinflammatory 15-epi-lipoxin A4 and inhibits thromboxane in a randomized human trial. Proc Natl Acad Sci USA 101:15178–15183
Morris T, Stables M, Hobbs A et al (2009) Effects of low-dose aspirin on acute inflammatory responses in humans. J Immunol 183:2089–2096
Kim SJ, Tominaga T (1989) Formation of lipoxins by the brain: Ischemia enhances production of lipoxins. Ann N Y Acad Sci 559:461–464
Sobrado M, Pereira MP, Ballesteros I et al (2009) Synthesis of lipoxin A4 by 5-lipoxygenase mediates PPARγ-dependent, neuroprotective effects of rosiglitazone in experimental stroke. J Neurosci 29:3875–3884
Decker Y, McBean G, Godson C (2009) Lipoxin A4 inhibits IL-1β-induced IL-8 and ICAM-1 expression in 1321N1 human astrocytoma cells. Am J Physiol Cell Physiol 296:C1420–1427
Kure I, Nishiumi S, Nishitani Y et al (2010) Lipoxin A4 reduces lipopolysaccharide-induced inflammation in macrophages and intestinal epithelial cells through inhibition of nuclear factor-κB activation. J Pharmacol Exp Ther 332:541–548
Morri H, Ozaki M, Watanabe Y (1994) 5′-flanking region surrounding a human cytosolic phospholipase A2 gene. Biochem Biophys Res Commun 205:6–11
Tanabe T, Tohnai N (2002) Cyclooxygenase isozymes and their gene structures and expression. Prostaglandins Other Lipid Mediat 68–69:95–114
Antonio V, Brouillet A, Janvier B et al (2002) Transcriptional regulation of the rat type IIA phospholipase A2 gene by cAMP and interleukin-1β in vascular smooth muscle cells: interplay of the CCAAT/enhancer binding protein (C/EBP), nuclear factor-κB and Ets transcription factors. Biochem J 368:415–424
Wientjes MG, Levy G (1988) Nonlinear pharmacokinetics of aspirin in rats. J Phamacol Exp Ther 245:809–815
Hauss-Wegrzyniak B, Willard LB, Del Soldato P et al (1999) Peripheral administration of novel anti-inflammatories can attenuate the effects of chronic inflammation within the CNS. Brain Res 815:36–43
Whitehead SN, Bayona NA, Cheng G et al (2007) Effects of triflusal and aspirin in a rat model of cerebral ischemia. Stroke 38:381–387
Ishizuka T, Niwa A, Tabuchi M et al (2008) Acetylsalicylic acid provides cerebrovascular protection from oxidant damage in salt-loaded stroke-prone rats. Life Sci 82:806–815
Wahner AD, Bronstein JM, Bordelon YM et al (2007) Nonsteroidal anti-inflammatory drugs may protect against Parkinson disease. Neurology 69:1836–1842
Ketterer MW, Brymer J, Rhoads K et al (1996) Is aspirin, as used for antithrombosis, an emotion-modulating agent? J Psychosom Res 40:53–58
Stolk P, Souverein PC, Wilting I et al (2010) Is aspirin useful in patients on lithium? A pharmacoepidemiological study related to bipolar disorder. Prostaglandins Leukot Essent Fatty Acids 82:9–14
Laan W, Grobbee DE, Selten JP et al (2010) Adjuvant aspirin therapy reduces symptoms of schizophrenia spectrum disorders: results from a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry 71:520–527
Doorduin J, de Vries EF, Willemsen AT et al (2009) Neuroinflammation in schizophrenia-related psychosis: a PET study. J Nucl Med 50:1801–1807
Acknowledgments
This work was supported entirely by the Intramural Research Program of the National Institute on Aging, NIH. None of the authors has a financial or other conflict of interest related to this work.
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article is available at http://dx.doi.org/10.1007/s11064-015-1583-4.
This article has been retracted on request of the Editor-in-Chief. The National Institutes of Health (NIH) has found that Dr. Mireille Basselin engaged in research misconduct by fabricating and/or falsifying data in "Basselin M, Ramadan E, Chen M, Rapoport SI, Anti-inflammatory effects of chronic aspirin on brain arachidonic acid metabolites. Neurochemical Research 36 (1) 139-145, 2011." Figure 2 A-E was falsified. Please note, none of the other authors are implicated in any way. Each of the co-authors of the manuscript has agreed to this retraction.
About this article
Cite this article
Basselin, M., Ramadan, E., Chen, M. et al. RETRACTED ARTICLE: Anti-Inflammatory Effects of Chronic Aspirin on Brain Arachidonic Acid Metabolites. Neurochem Res 36, 139–145 (2011). https://doi.org/10.1007/s11064-010-0282-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-010-0282-4