
Abstract
Galectin-1, an endogenous mammalian lectin, has been implicated in a variety of CNS disorders. However, its role in cerebral ischemia is still elusive. In the present study, we investigated the effect of recombinant galectin-1 on production of astrocytic brain-derived neurotrophic factor (BDNF) and functional recovery following ischemia. Endogenous galectin-1 was found to be markedly upregulated, paralleled with increased astrocytic BDNF production under ischemic conditions both in vitro and in vivo. Administration of galectin-1significantly enhanced the expression and secretion of astrocytic BDNF in dose dependent manner. Moreover, rats subjected to photochemical cerebral ischemia showed reduced neuronal apoptosis in ischemic boundary zone and improved functional recovery after brain infusion of galectin-1 (1 μg/days, 7 days). These results suggest that induction of BDNF in astrocytes by galectin-1 may be a promising intervention to attenuate brain damage after stroke.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In central nervous system (CNS), astrocytes serve many housekeeping functions essential for neuronal function [1, 2]. It is also actively involved in pathological reactions to all forms of CNS insults, such as stroke, Alzheimer Disease, epilepsy and infectious diseases [3–5]. Under ischemic conditions, astrocytes become reactive and carry out critical functions such as maintenance of ionic homeostasis, prevention of excitotoxicity, scavenging free radicals, provision of nutrients and growth factors, promotion of neovascularization, and support of synaptogenesis and neurogenesis that may influence the outcome of ischemic injury [6, 7]. However, the role of astrocyte in the process of neuronal damage induced by ischemia remains to be determined.
Galectin-1 (Gal-1) is a soluble carbohydrate-binding protein that has been implicated in a variety of biological events [8–11]. In CNS, previous findings show that it is widely expressed in both neurons and glia [12, 13] and has general effects after injuries. It has been reported to be involved in neurite outgrowth and synaptic connectivity of peripheral nerve, resistance to glutamate toxicity and cellular adaptation to redox status after stroke, proliferation of neural stem cells and cerebellar astrocytes, as well as production of inflammatory and neurotrophic factors after injuries [8, 11, 12, 14, 15]. Those studies suggest that Gal-1 may have therapeutic potential to prevent neuronal damage after stroke.
In the present study, we investigated the temporal expression of Gal-1 in CNS cells after ischemia. Moreover, the effect of recombinant Gal-1 on astrocytic expression of BDNF, neuronal loss and functional recovery of rats post-ischemia was also studied. Our results show that Gal-1 was mainly expressed by activated astrocytes after ischemia, and exogenous Gal-1 could enhance the expression and secretion of BDNF from cortical astrocytes in a dose dependent manner. Furthermore, Gal-1 treatment suppressed the neuronal death and improved the functional recovery of rats following ischemia.
Experiment Procedure
Cell Cultures and Anoxic Treatment
Cultures of astrocytes from neonatal rat cortex were prepared according to the method described previously [16]. Cells were plated in culture flasks at a density of 1–3 × 106/ml and maintained at 37°C and 5%CO2/20%O2 in DMEM-F12 supplemented with 10% FBS and 10% NBS (Hyclone). In the second week of maintenance in vitro, non-astroglial cells were removed by shaking. The majority of remaining cells were astrocytes. Primary astrocyte cultures were trypsinized and re-plated at 3 × 105 cells/cm2 onto new flask or poly-l-lysine coated glass coverslips. The medium was changed every 3 days until confluence.
Purity of astrocyte cultures was determined by double-labeling of cultured cells with anti-GFAP (Neomarkers, 1:200) and DAPI (Sigma, 10 μg/ml). It indicated that more than 99% of the confluent cells were GFAP(+) astrocytes.
In order to mimic ischemic injury in vitro, anoxic treatment of astrocytes was introduced as our previous report [16]. Briefly, cells were cultured with serum-free DMEM-F12 media in cell incubator (Thermo scientific) at 37°C, which was flushed with a gas mixture of 93% N2/5% CO2/2% O2.
Photochemical Local Cerebral Ischemia Model
Adult male Wistar rats weighing 220–280 g were used. The experimental procedures were performed in accordance with protocols approved by the Governmental Animal Care Committee at Tongji Medical College. Rats were randomly assigned to sham-operated (n = 6), ischemia (n = 30) and Gal-1 treated (n = 18) groups. Rats were anesthetized with intraperitoneal ketamine (75 mg/kg) and xylazine (20 mg/kg) injection. Using aseptic technique, 2 cm longitudinal incision was made over the scalp and the periosteum was removed. Rose Bengal (Sigma, 10 mg/kg, dissolved in 0.9% NaCl) was injected i.p. Sham-operated mice were injected with 0.9% NaCl. Then illumination was started with a cold light source for 20 min to induce focal cerebral ischemia (KL1500LCD, Schott. 15 W, 550 nm) [17]. Rats in vehicle groups were sacrificed at days 1, 3, 7, 15 and 30 post-ischemia, respectively, whereas those in Gal-1 treated groups were killed at days 7, 15 and 30.
Gal-1 Intervention
Gal-1(R&D systems) was dissolved in 0.9% saline with 1 mg/ml BSA (Hyclone) and 250 μM β-mercaptoethanol. Gal-1 free solution was used as control. In astrocyte cultures, Gal-1 was applied in culture media at the beginning of anoxia with three concentrations, 10, 1 or 0.1 μg/ml.
For animal treatment, Gal-1 was dissolved in 0.9% saline. Five microliters Gal-1 (1 μg/days, 7 days) or saline was infused into the cerebellomedullary cistern through a catheter in 5 min by microsyringe [18]. In order to keep its stability and activity, Gal-1 was given immediately after the operation and followed by daily injection as described in previous reports [19].
Western Blot Analysis
Cell cultures were quickly scraped off and homogenized by sonication in RIPA Lysis Buffer [20] with protease inhibitor cocktail (Sigma). Lysates were then centrifuged at 12,000g (4°C) for 10 min and the supernatant was collected for protein concentration assay using a BCA kit [20].
Samples containing 60 μg total protein were loaded on 15% SDS–PAGE. After transferring to membrane, the unspecific binding was blocked by a buffer containing 5% fat-free dry milk in TBS. The membrane was then incubated with primary antibody diluted in blocking buffer overnight at 4°C. After washing membranes with 0.1% Tween-20-TBS 10 min for four times, it was incubated with horseradish peroxidase-conjugated IgG for 1 h at room temperature, and then visualized with ECL kit [20]. The following antibodies were used: goat anti-Gal-1 (R&D systems, 1:400), rabbit anti-BDNF (R&D systems, 1:500), mouse anti-β-actin (Neomarkers, 1:20,000) and horseradish peroxidase-conjugated anti- goat/rabbit/mouse IgG (Pierce, 1:2,500). The integrated optical density (OD) of signals was semi-quantified by Kodak Digital Science 1D system. After normalized with β-actin, values were displayed as relative amount to control (control was loaded as 100%).
Immunofluorescent and TUNEL Staining
Animals were sacrificed under anesthesia and perfused with saline transcardially, followed by ice-cold 4% paraformaldehyde postfixation for additional 4 h. Brains were put in 30% sucrose-PBS at 4°C for 3 days, and cut at 30 μm thickness. Primary astrocyte cultures on glass coverslips were fixed with methanol for 10 min at −20°C, then blocked with 10% BSA-0.25%Triton-PBS.
Subsequently, slices were treated with primary antibody for 18 h at 4°C, visualized with secondary antibody for 1 h at room temperature, and observed with Laser scanning confocal microscope (Olympus). The following antibodies were used: goat anti-Gal-1 (1:200), rabbit anti-BDNF (1:200), mouse anti-GFAP (1:200), mouse anti-CD11b/c (BD biosciences, 1:50), mouse anti-CNPase (Neomarkers, 1:200), Cy3-conjugated anti-goat/rabbit IgG and FITC-conjugated anti-goat/mouse IgG (Pierce, 1:200).
DNA Fragmentation Analysis with TdT-Mediated dUTP Nick-End Labeling (TUNEL) was performed using the Red In situ Apoptosis Detection Kit (Chemicon) strictly according to the manufacturer’s instructions. The slices were then co-stained with NeuN (Chemicon, 1:200). Site near the bottom of the infarct was captured by microscope (Olympus) and analyzed with Microimage analysis software (NIH image).
Since there is no acceptable definition of ‘ischemic boundary zone’ [21], we observed the area 300 μm below the infarct, where evident astrogliosis can be seen especially at 7 days post-ischemia. Six rats were randomly enrolled in each group and four sections from the core of ischemic site were taken for labeling. The number of positive cells in 300 μm wide around the margin of injury site was counted at high-power magnification (×200). GFAP and BDNF, or GFAP and Gal-1 double-stained cells were counted regardless the intensity of labeling. Cell counts were performed by two different investigators who were blind to the classification of tissues. The estimated cell number (n) was the average of values from four adjacent sections.
Behavioral Measurements
Behavioral changes of 6 rats in either vehicle or Gal-1 treated group were evaluated according to a modified Neurological Severity Score (NSS) [22]. Tests were performed before and at days 1, 7, 15 and 30 post-ischemia by an investigator blinded to experimental groups. The neurological score was graded on a scale of 0–10 (Normal score = 0, maximal deficit score = 10), which was composed of motor, sensory, and reflex tests (Table 1). Scores of 8–10 indicate severe injury; 4–6, moderate injury; and 1–3, middle injury.
Statistical Analysis
Data are presented in mean ± S.E. Statistically significant differences (defined as P < 0.05) were evaluated by one-way ANOVA followed by Tukey’s post hoc test or Student’s t test.
Results
Gal-1 is Increased in Astrocyte Cultures Under Anoxic Conditions
Under normal conditions, astrocytes were initially identified according to their characteristic morphological shapes which were small in soma size, appearing either as round or irregular shapes with visible primarily processes extending from the soma. Gal-1 was shown to be moderately expressed in cultured astrocytes mostly in cytoplasm (Fig. 1A1). After anoxic treatment, astrocytes were activated and became hyperplasia and hypertrophy, which was accompanied by elevated fluorescent density of Gal-1 (Fig. 1A2).

Expression of Gal-1 in astrocyte cultures following anoxia. (A) Double staining of GFAP and Gal-1. It demonstrated that Gal-1 was immunoreactive in astrocytes (A1) and was upregulated after astrocyte activation induced by 6 h anoxia (A2). Scale bar = 50 μm. (B) Expression of Gal-1 protein after anoxia. It showed that expression of astrocytic Gal-1 was elevated at 6 and 12 h, but decrease at 3 days post-anoxia, compared to normal cultured control. Values are expressed as the mean ± S.E. (n = 6). # P < 0.01 (one-way ANOVA test)
Corresponding to immuocytochemical staining, western blots displayed the temporal changes of Gal-1 expression in cultured astrocytes after anoxia. As shown in Fig. 1B, Gal-1 expression was increased significantly at 6 h and 12 h post-anoxia, and decreased gradually later.
Gal-1 Enhances Astrocytes to Produce BDNF After Anoxia In Vitro
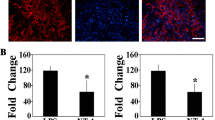
BDNF is a neuroprotective polypeptide essential for neuron function. Astrocytes can produce several neurotrophic factors including BDNF to maintain neuronal activity under both physiological and pathological conditions. As shown in Fig. 2A1, BDNF was expressed in cultured astrocytes. Although BDNF immunoreactivity was weak at 1 day anoxia (Fig. 2A2), addition of Gal-1 increased the expression of BDNF significantly (Fig. 2A3).

Gal-1 treatment enhances expression of BDNF in astrocyte cultures after anoxia. (A) BDNF immunoreactivity in astrocytes normal-cultured (A1), after 1 day anoxia (A2) and treated by 10 μg/ml Gal-1 (A3). Scale bar = 100 μm. (B) Expression of BDNF protein in astrocytes after 1 day treatment. (C) Concentration of BDNF in supernatant detected by ELISA after 1 day treatment. It exhibited that expression and secretion of astrocytic BDNF were enhanced by Gal-1 dose-dependently. Values are expressed as the mean ± S.E. (n = 6). # P < 0.01; * P < 0.05 vs. anoxia group (one-way ANOVA test)
Western blot analysis revealed that the expression of BDNF was upregulated by Gal-1 treatment in a dose-dependent manner (Fig. 2B). At 1 day anoxia, 1 and 10 μg/ml Gal-1 elevated BDNF expression in astrocytes significantly, compared to vehicle treated group (Fig. 2C). We next measured the BDNF secreted into cultured media by ELISA analysis (Fig. 2D). Consistent with the expression in cells, Gal-1 enhanced astrocytic secretion of BDNF dose-dependently after the exposure (506.60 ± 46.24 pg/ml in 10 μg/ml Gal-1 treated group vs. 362.61 ± 20.38 pg/ml of anoxia group).
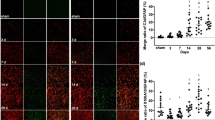
Expression of Gal-1 and BDNF are Paralleled in Activated Astrocytes in Ischemic Boundary Zone
We next tested whether Gal-1 and BDNF was expressed in vivo and how ischemia affected their expression. The region as shown in Fig. 3A was focused for investigation. Immunofluorescent staining showed that Gal-1 was expressed in sham-operated brain (Fig. 3B1) and upregulated in activated astrocytes after ischemia (Fig. 3B2–4). By labeling Gal-1 with markers special for neuron (Fig. 4A), astrocyte (Fig. 3B), microglia (Fig. 4B) and oligodendrocyte (Fig. 4C), the expression of Gal-1 was found to mainly co-localize with GFAP(+) astrocytes in ischemic boundary zone. Statistic analysis showed that percentage of Gal-1(+) astrocytes peaked at 7 days post-ischemia (Fig. 3D). Meanwhile, only transient upregulation of Gal-1 expression was found in cortical neurons at 6 h post-ischemia (Fig. 4A2), whereas few Gal-1(+) microglia and oligodendrocyte was detected either in normal cortex (Fig. 4B1, C1) or after the injury (Fig. 4B2, C2).

Expression of BDNF and Gal-1 in astrocytes in rats post-ischemia. (A) Schematic drawing of coronal brain sections taken from ischemic animals induced by photochemistry. Infarct (dark gray) in the cortex is surrounded with ischemic boundary zone, where we took photos for analysis. (B/C) Double staining of Gal-1/BDNF and GFAP in cortex of sham-operated or ischemic animals. (D) Time profile of astrocytic Gal-1/BDNF expression post-ischemia after statistic analysis. It demonstrated that BDNF was elevated post-ischemia, paralleling with the increase of Gal-1, both of which were localized in activated astrocytes. # P < 0.01, * P < 0.05 vs. sham-operated control (one-way ANOVA test). E1 and E2: BDNF expression in activated astrocytes after Gal-1 infusion (1 μg/day, 7 days) at days 7 and 15 post-ischemia, respectively. (F) Comparison of BDNF (+) astrocyte rates between Gal-1 and vehicle treated groups. After 7 days Gal-1 treatment, astrocytic BDNF expression was enhanced significantly. # P < 0.01, * P < 0.05 vs. vehicle treated control (Student’s t test). Values are expressed as the mean ± S.E. (n = 6). Scale bar = 100 μm

Expression of Gal-1 in neuron, microglia and oligodendrocyte in rat cerebral cortex. Gal-1 was double-stained with NeuN, CD11b/c and CNPase, the cell markers for neuron, microglia and oligodendrocyte, respectively. By time dependent observation, little Gal-1 immunoreactivity was seen in neuron (A1), microglia (B1) and oligodendrocyte (C1) of sham-operated rats. Although transient expression appeared in neurons around ischemic infarct at 6 h post-ischemia (A2), no Gal-1 immunoreactivity was detected in activated microglias and oligodendrocytes, taking 3 days post-ischemia for example (B2 and C2). Scale bar = 100 μm
In sham-operated animals, BDNF was mainly expressed in neuron-like cells. Contrastively, only weak immunoreactivity was found to merge with GFAP(+) cells (Fig. 3C1). Consistent with Gal-1, the expression of BDNF was also upregulated after ischemia, which preferentially appeared in activated astrocytes (Fig. 3C2–4). Percentage of BDNF(+) astrocytes peaked at 7 days post-ischemia (63.34 ± 6.33% vs. 8.88 ± 1.56% of sham-operated group) (Fig. 3D).
Gal-1 Enhances Expression of BDNF in Astrocytes in Ischemic Boundary Zone
To investigate whether Gal-1 enhances the production of BDNF in vivo, we performed brain infusion of Gal-1 and studied the expression of astrocytic BDNF by immunofluorescent staining. Significant cytoplastic location of BDNF immunoreactivity appeared in activated astrocytes after the treatment at days 7 and 15 post-ischemia (Fig. 3E1, E2), which was accompanied by the elevation of BDNF(+) astrocyte rates (Fig. 3F).
Gal-1 Attenuates Neuronal Loss and Improves Functional Outcome of Rats Suffering Ischemia
Since Gal-1 treatment enhanced the production of BDNF, it is reasonable to conceive that it can attenuate ischemic neuronal death and provide neuroprotective effects. Thus we performed double staining of NeuN and TUNEL to investigate the neuronal death induced by ischemia (Fig. 5A). TUNEL(+) neurons were seen as early as 1 day, increased at 3 days and peaked at 7 days post-ischemia. Contrastively, Gal-1 treatment significantly reduced the percentage of TUNEL(+) neurons (day 7, 13.48 ± 3.31% vs. 32.14 ± 4.64%; day 15, 11.91 ± 3.23% vs. 28.14 ± 4.63% of vehicle treated groups) (Fig. 5B).

Gal-1 attenuates neuronal apoptosis and improves behavioral outcomes after ischemia. (A) Double staining of NeuN and TUNEL in cortex of sham-operated and 7 days ischemic rats. (B) Cartogram for statistic results of neuronal apoptosis. Suppression of neuronal apoptosis was seen after 7 days Gal-1 infusion both at days 7 and 15 post-ischemia. (C) Statistic results of Neurological severity score (NSS). It showed that persistent sensorimotor damage appeared quickly after the ischemia, with the maximum score at 1 day post-ischemia. Contrastively, Gal-1 infusion promoted functional recovery effectively from 7 days post-ischemia on. Values are expressed as the mean ± S.E. (n = 6). # P < 0.01, * P < 0.05 vs. vehicle-treated animals (Student’s t-test). Scale bar = 100 μm
In addition, administration of Gal-1 post-ischemia significantly mitigated the functional deficits during 1 month observation (Fig. 5C). Although all rats exhibited obvious functional deficits at 1 day post-injury, improvement in behavioral outcome was observed within 7 days in both Gal-1 and vehicle-treated rats. By application of Gal-1, it resulted in progressively lower final neurological severity scores compared to that of vehicle treated groups, which is in accordance with the reduction of TUNEL(+) neurons in ischemic boundary zone.
Discussion
Our present study demonstrates that Gal-1 is an endogenous protein that can be upregulated in activated astrocytes after ischemic injuries, and exogenous Gal-1 treatment induces the production of BDNF from cortical astrocytes in vitro and vivo, which may contribute to the recovery of focal cerebral ischemia, although we are unable to conclude that the functional recovery is directly benefited from the increased astrocytic BDNF.
Gal-1 is a ubiquitously expressed 14.5 kDa protein which can be localized in the developing brain, olfactory system, and restricted to peripheral nervous tissues with maturation [11, 12, 23]. However, the expression of Gal-1 in CNS is still never completely identified. Previous studies showed that it is expressed in cell body of neurons in several brain stem nucleuses and astrocytes from subventricular zone and cerebellum [12, 13, 24]. After injuries, Gal-1 expression seems to tightly correlate with activated astrocytes. It was reported to be upregulated in subventricular zone astrocytes and contributed to adult neurogenesis after focal ischemias [12]. After systemic kainate administration, Gal-1 is remarkably increased in activated astrocytes of the CA3 subregion and dentate gyrus of hippocampus [25]. In this study, we found that Gal-1 expression was highly induced in cortical astrocytes activated by either anoxia to cell cultures or photochemical thrombosis in animals. Collectively, these documents suggest that Gal-1 is mainly expressed by astrocytes in CNS.
Until recently, it’s still not thoroughly known that how astrocytic Gal-1 functions in brain after ischemia. Previous studies prompt that astrocytic Gal-1 likely act as an endogenous molecule for certain functions following injuries, maybe as a growth-stimulating or survival factor for neurons [26, 27] and a chemical mediator of inflammatory response [28]. It also has been confirmed that after 1-day treatment to cerebellar astrocytes with Gal-1, production of BDNF was greatly increased [27].
We here observed the Gal-1 increased the expression of BDNF in cortical astrocytes, which were directly involved in focal cerebral ischemia. Different to the report by Sasaki et al. [27], BDNF expression induced by Gal-1 in cortical astrocytes is not fiercely as that in cerebullar astrocytes. It may be partly attributed to different models used and heterogeneity of astrocytes from different brain regions [29]. Considering of Gal-1’s lectin activity that may induce agglutination of red blood cells [30], we adopted a dose no higher than 10 μg/ml (about 10 times lower than the report). Through dose-dependant analysis, 10 μg/ml Gal-1 was found to be effective without significant side-effects.
Intracellular Gal-1 was reported to be a downstream target of △FosB regulating neuronal fate [31], while endogenous Gal-1 is associated with cell multiplication [10, 25]. Studies have showed that secreted endogenous Gal-1 can act on the relevant receptors and induce downstream effects [8]. We here showed that endogenous Gal-1 can be induced by anoxia, and exogenous Gal-1 contributed to the astrocytic BDNF production. It remains to be determined whether exogenous and endogenous Gal-1 acts in different way.
BDNF is a traditional neurotrophic factor which is sensitive to various stimuli [32, 33]. Although BDNF was mainly produced by neurons in physical condition [32], our results support that increased BDNF after ischemia mainly comes from activated astrocytes, and can be elevated by agents like Gal-1. The enhanced BDNF after injuries was reported to rescue damaged neurons and promote axonal outgrowth, remyelination and regeneration, thus take a neuroprotective and neurorestorative role after insults [22, 34, 35]. Those functions are proposed to be fulfilled in activity-dependent secretion, which aids its local diffusion in brain milieu [36]. Accordingly, we found that secretion of BDNF was also enhanced by Gal-1 treatment.
Because of its role in enhancing BDNF production, Gal-1 is supposed to have neuroprotective potential after ischemia. We evaluated its effect in a rat model of focal ischemia induced by photochemical injury, which has been employed to produce reproducible pathological features and the main characteristic stages of stroke [37]. The consequent infarct in cortical regions facilitated our study of structural and behavioral consequences of ischemia. In this model, we found that apoptotic neurons were focused around the infarct and peaked at 7 day post-ischemia. After 7-days continual infusion of Gal-1 into subarachnoid space, percentage of TUNEL(+) neurons was significantly reduced. Further neurological assessment showed that Gal-1 treatment caused long-term improvement in neurological function. This result was in agreement with a previous report on gerbil focal ischemia model showing that Gal-1 promotes functional recovery after ischemia through facilitating neurogenesis [10].
We would like to elucidate that recovery of ischemic rats might come from multiple effects of several pathways because Gal-1 takes a general role in CNS [8, 10, 38]. Although dependable data about the dose of astrocytic BDNF valuable for neuroprotection has not been identified, the increased production of astrocytic BDNF may partly mediate this neuroprotective effect at least. Whatever, our discoveries suggest that Gal-1 may be a candidate for the therapy of cerebral ischemia.
References
Mazzanti M, Sul JY, Haydon PG (2001) Glutamate on demand: astrocytes as a ready source. Neuroscientist 7:396–405
Abbott NJ (2002) Astrocyte-endothelial interactions and blood-brain barrier permeability. J Anat 200:629–638
Louw DF, Masada T, Sutherland GR (1998) Ischemic neuronal injury is ameliorated by astrocyte activation. Can J Neurol Sci 25:102–107
Meda L, Baron P, Scarlato G (2001) Glial activation in alzheimer’s disease: the role of abeta and its associated proteins. Neurobiol Aging 22:885–893
Seifert G, Carmignoto G, Steinhauser C (2010) Astrocyte dysfunction in epilepsy. Brain Res Rev 63(1–2):212–221
Swanson RA, Ying W, Kauppinen TM (2004) Astrocyte influences on ischemic neuronal death. Curr Mol Med 4:193–205
Pekny M, Wilhelmsson U, Bogestal YR et al (2007) The role of astrocytes and complement system in neural plasticity. Int Rev Neurobiol 82:95–111
Lekishvili T, Hesketh S, Brazier MW et al (2006) Mouse galectin-1 inhibits the toxicity of glutamate by modifying nr1 nmda receptor expression. Eur J Neurosci 24:3017–3025
Kato T, Ren CH, Wada M et al (2005) Galectin-1 as a potential therapeutic agent for amyotrophic lateral sclerosis. Curr Drug Targets 6:407–418
Ishibashi S, Kuroiwa T, Sakaguchi M et al (2007) Galectin-1 regulates neurogenesis in the subventricular zone and promotes functional recovery after stroke. Exp Neurol 207:302–313
Gaudet AD, Steeves JD, Tetzlaff W et al (2005) Expression and functions of galectin-1 in sensory and motoneurons. Curr Drug Targets 6:419–425
Sakaguchi M, Imaizumi Y, Okano H (2007) Expression and function of galectin-1 in adult neural stem cells. Cell Mol Life Sci 64:1254–1258
Akazawa C, Nakamura Y, Sango K et al (2004) Distribution of the galectin-1 mrna in the rat nervous system: its transient upregulation in rat facial motor neurons after facial nerve axotomy. Neuroscience 125:171–178
Almkvist J, Karlsson A (2004) Galectins as inflammatory mediators. Glycoconj J 19:575–581
Endo T (2005) Glycans and glycan-binding proteins in brain: galectin-1-induced expression of neurotrophic factors in astrocytes. Curr Drug Targets 6:427–436
Zhu Z, Zhang Q, Yu Z et al (2007) Inhibiting cell cycle progression reduces reactive astrogliosis initiated by scratch injury in vitro and by cerebral ischemia in vivo. Glia 55:546–558
Dietrich WD, Prado R, Pravia C et al (1999) Delayed hypovolemic hypotension exacerbates the hemodynamic and histopathologic consequences of thromboembolic stroke in rats. J Cereb Blood Flow Metab 19:918–926
Takemoto Y (1993) Regional vasoconstriction and excessive grooming induced by l-arginine injection into the cisterna magna of conscious rats. Jpn J Physiol 43:389–402
Sakaguchi M, Shingo T, Shimazaki T et al (2006) A carbohydrate-binding protein, galectin-1, promotes proliferation of adult neural stem cells. Proc Natl Acad Sci USA 103:7112–7117
Hoane MR, Pierce JL, Kaufman NA et al (2008) Variation in chronic nicotinamide treatment after traumatic brain injury can alter components of functional recovery independent of histological damage. Oxid Med Cell Longev 1:46–53
Sofroniew MV (2009) Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci 32:638–647
Schabitz WR, Berger C, Kollmar R et al (2004) Effect of brain-derived neurotrophic factor treatment and forced arm use on functional motor recovery after small cortical ischemia. Stroke 35:992–997
Regan LJ, Dodd J, Barondes SH et al (1986) Selective expression of endogenous lactose-binding lectins and lactoseries glycoconjugates in subsets of rat sensory neurons. Proc Natl Acad Sci U S A 83:2248–2252
Yang JW, Kang SU, Engidawork E et al (2006) Mass spectrometrical analysis of galectin proteins in primary rat cerebellar astrocytes. Neurochem Res 31:945–955
Kajitani K, Nomaru H, Ifuku M et al (2009) Galectin-1 promotes basal and kainate-induced proliferation of neural progenitors in the dentate gyrus of adult mouse hippocampus. Cell Death Differ 16:417–427
McGraw J, Oschipok LW, Liu J et al (2004) Galectin-1 expression correlates with the regenerative potential of rubrospinal and spinal motoneurons. Neuroscience 128:713–719
Sasaki T, Hirabayashi J, Manya H et al (2004) Galectin-1 induces astrocyte differentiation, which leads to production of brain-derived neurotrophic factor. Glycobiology 14:357–363
Rabinovich GA, Sotomayor CE, Riera CM et al (2000) Evidence of a role for galectin-1 in acute inflammation. Eur J Immunol 30:1331–1339
Hewett JA (2009) Determinants of regional and local diversity within the astroglial lineage of the normal central nervous system. J Neurochem 110:1717–1736
Gupta RK, Pande AH, Gulla KC et al (2006) Carbohydrate-induced modulation of cell membrane. Viii. Agglutination with mammalian lectin galectin-1 increases osmofragility and membrane fluidity of trypsinized erythrocytes. FEBS Lett 580:1691–1695
Miura T, Ohnishi Y, Kurushima H et al (2005) Regulation of the neuronal fate by deltafosb and its downstream target, galectin-1. Curr Drug Targets 6:437–444
Ferrer I, Krupinski J, Goutan E et al (2001) Brain-derived neurotrophic factor reduces cortical cell death by ischemia after middle cerebral artery occlusion in the rat. Acta Neuropathol 101:229–238
Murer MG, Yan Q, Raisman-Vozari R (2001) Brain-derived neurotrophic factor in the control human brain, and in alzheimer’s disease and parkinson’s disease. Prog Neurobiol 63:71–124
Li L, Xu Q, Wu Y et al (2003) Combined therapy of methylprednisolone and brain-derived neurotrophic factor promotes axonal regeneration and functional recovery after spinal cord injury in rats. Chin Med J (Engl) 116:414–418
Pezet S, Malcangio M (2004) Brain-derived neurotrophic factor as a drug target for cns disorders. Expert Opin Ther Targets 8:391–399
Thomas K, Davies A (2005) Neurotrophins: a ticket to ride for bdnf. Curr Biol 15:R262–R264
Watson BD, Dietrich WD, Busto R et al (1985) Induction of reproducible brain infarction by photochemically initiated thrombosis. Ann Neurol 17:497–504
Horie H, Kadoya T (2004) Galectin-1 plays essential roles in adult mammalian nervous tissues. Roles of oxidized galectin-1. Glycoconj J 19:479–489
Acknowledgments
This work was supported by China National Funds for Distinguished Young Scientists (30725019), National Natural Science Foundation (30800341, 30971007, 30800340) and Medical Research Project of Henan (WKJ-2007-2-031).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Wen-sheng Qu and Yi-hui Wang contributed equally to this work.
Rights and permissions
About this article
Cite this article
Qu, Ws., Wang, Yh., Wang, Jp. et al. Galectin-1 Enhances Astrocytic BDNF Production and Improves Functional Outcome in Rats Following Ischemia. Neurochem Res 35, 1716–1724 (2010). https://doi.org/10.1007/s11064-010-0234-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-010-0234-z