
Abstract
Hydrogen sulfide (H2S) is a gaseous messenger and serves as an important neuromodulator in the central nervous system. The current study was undertaken to investigate whether H2S attenuates the neuronal injury induced by vascular dementia (VD). Rats were subjected to bilateral common carotid artery and vertebral artery occlusion for 5 min three times in an interval of 5 min to induce VD. An H2S donor, sodium hydrosulfide (NaHS) or an inhibitor of cystathionine-β-synthase, hydroxylamine (HA) was administered intraperitoneally. The number of neurons in the hippocampus was determined by hematoxylin and eosin staining, and the performance of learning and memory was tested by the Morris water maze. H2S content in plasma was evaluated. Apoptosis in the hippocampus was assessed by flow cytometry. In addition, Bcl-2 and Bax expression was analyzed by immunohistochemical staining. The neuronal injury occurred gradually with a decreased number of neurons and increased apoptosis ratio in the hippocampus over 720 h after VD. The H2S level was also gradually decreased in plasma over 720 h after VD, which negatively correlated with the apoptosis ratio in the hippocampus after VD. In addition, NaHS treatment significantly attenuated neuronal injury and improved neural functional performance, whereas HA exaggerated the neuronal injury and exacerbated learning and memory at 720 h after VD. Furthermore, NaHS treatment markedly improved the ratio of Bcl-2 over Bax with increased Bcl-2 expression and decreased Bax expression. In contrast, HA reduced the ratio of Bcl-2 over Bax. It is suggested that H2S attenuates VD injury via inhibiting apoptosis and may have potential therapeutic value for VD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
With an increase of elderly population, aging-related diseases such as hypertension, arteriosclerosis and different forms of dementia are also increased [1]. One of these diseases is vascular dementia (VD). VD has already become a major public problem, which is characterized by histopathological damage and progressive intellectual decline produced by ischemic hypoxia or hemorrhage brain lesion [2–4]. It represents the second most common dementia accounting for about a quarter to a half of all cases of dementia in developed countries [2–4]. Unfortunately, the pathogenesis of the cognitive decline is not fully understood, and there are no known effective treatments or preventive strategies for VD yet.
Hydrogen sulfide (H2S) has been best known as a toxic environmental pollutant emerging from sewers, marshes, and volcanic eruptions as a toxic gas [5]. But recent studies have suggested the gas’s improving reputation over the past two decades [6, 7]. There is now accumulating evidence that it is an endogenously produced gaseous messenger and in particular, serves as an important neuromodulator in the central nervous system [8]. In mammalian tissues, two pyridoxal-5′-phosphate-dependent enzymes, cystathionine-β-synthase (CBS) and cystathionine-γ-lyase (CSE) are responsible for most of the biosynthesis of H2S from l-cysteine [9]. The endogenous production of H2S was initially described in the brain by Abe in 1996 [8]. CBS, which is highly expressed in the hippocampus and cerebellum, is a predominant source of H2S in the central nervous system whereas CSE is a major H2S-producing enzyme in the cardiovascular system [10].
Some studies reported that H2S promotes vascular smooth muscle relaxation and induces vasodilation of isolated blood vessels including aorta, mesenteric artery and pulmonary artery [11–14]. H2S has been shown to inhibit leukocyte-endothelial cell interactions in vivo indicating an anti-inflammatory action [15]. H2S inhibits superoxide formation, NOX-1 expression and reactivity in human vascular smooth muscle cells [16] and endothelial cells [17] and protects neurons from oxidative stress [18]. H2S enhances the reducing activity in neurons, which may lead to a neurotrophic role in the brain [19]. Some reports have suggested that H2S exerts a protective effect against ischemia reperfusion injury in heart [20], liver [21] and lung [22]. However, the protective effect of H2S has not been evaluated in VD induced by cerebral ischemia reperfusion. Some evidence suggests that apoptosis could be a mechanism of VD [23, 24]. Interestingly, the anti-apoptotic effect of H2S has also been demonstrated in vitro [25] and in vivo [20, 21, 26], which may promote cytoprotection from VD injury. Therefore, in the present study, we determined if VD induced apoptosis and the change of H2S level in the plasma and the hippocampus and evaluated the effects of NaHS, an H2S donor or HA, a CBS inhibitor treatment on VD injury and investigated if the H2S reduced VD injury through suppressing apoptosis.
Experimental Procedure
Animals
Male Sprague-Dawley rats (300 ± 20 g) provided by The Experimental Animal Center of Hebei Medical University were used for the study. The rats were housed at a temperature of 23 ± 1°C with a 12 h light–dark cycle (light on 7 a.m.–7 p.m.), and had free access to the food and water. The rats were fasted for 12 h with free access to water before the experiment. Animal care and use conformed to guidelines for care and use of experimental animals. All experimental procedures described in this study were approved by the Animal Care and Use Committee at Hebei Medical University.
Reagents and Antibodies
Antibodies against Bcl-2 and Bax were purchased from Santa Cruz (Santa Cruz Biotechnology Inc., CA, USA). As an H2S donor, 14 μmol/kg sodium hydrosulfide (NaHS) was administered intraperitoneally 15 min prior to VD. This dose of NaHS is reported effective in protection against myocardial ischemia of rats [27]. When NaHS is dissolved in water, HS− is released and forms H2S with H+. This provides a solution of H2S at a concentration that is about 33% of the original concentration of NaHS [28]. About 5 μmol/kg hydroxylamine (HA) was administered intraperitoneally 15 min prior to VD as a CBS inhibitor. NaHS and HA were obtained from Sigma–Aldrich (Sigma–Aldrich, St. Louis, MO) and were dissolved in saline. Control rats received saline only.
VD Surgery Procedure
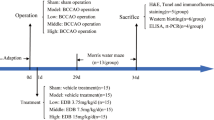
VD was induced by the 4 vessel-occlusion (4-VO) model. Briefly, after rats were anesthetized with choral hydrate (350 mg/kg, i.p.), a midline incision was made in the dorsal neck and the paraspinal muscles were separated from the middle. The alar foramina of atlas were identified, and the bilateral vertebral arteries through the alar foramina were electrocauterized to yield complete and permanent cessation of circulation. Twenty-four hours after the operation, bilateral common carotid arteries were isolated through a ventral midcervical incision, and occluded for 5 min three times in an interval of 5 min under ether anesthesia. Rectal temperature was continually monitored and maintained at 36.5–37.5°C with a heating blanket during VD surgery.
Measurement of H2S
Plasma for testing H2S content was obtained from the blood samples, which were collected from the abdominal aorta via a stainless steel needle into a heparinized syringe and immediately centrifuged. The measuring procedures are essentially described in the literature with modifications [29]. Briefly, plasma was mixed with trichloroacetic acid (10% w/v, 250 μl), zinc acetate (1% w/v, 250 μl), N,N-dimethyl-p-phenylenediamine sulphate (20 μM; 133 μl) in 7.2 M HCl and FeCl3 (30 μM; 133 μl) in 1.2 M HCl in parafilm-enveloped Eppendorf tubes. After 15 min, this mixture was centrifuged at 4,000×g for 10 min. The supernatant was collected and its absorbance measured in 96-well plates at a wavelength of 670 nm with a microplate reader (Elx-800; Bio-Tex). The concentration of H2S was calculated by using a standard H2S curve.
Morris Water Maze
Spatial learning and memory was tested according to the classic Morris protocol with modifications [30]. Briefly, the maze consisted of a circular tank, 1.2 m in diameter, filled with water and conceptually divided into four quadrants. Rats were trained to find a submerged escape platform, which was located in the center of one of the quadrants. Each rat was placed in the pool without the platform for 120 s to habituate to the environment on the day before trials. On days 1–4, rats were trained for 20 trials (five trials a day) to find the platform within 120 s. The time that rats spent in finding the platform is called escape latency. For each rat, the quadrant in which the platform was located remained constant, but the point of immersion in the pool varied among four quadrants in a random order for the 20 trials. On mounting the platform, the rats were given a 30-s rest period, after which the next trial was started. If the rat did not find the platform in 120 s, it was manually placed on the platform for a 30-s rest, and the escape latency was 120 s.
Flow Cytometric Analysis
The tissue from bilateral hippocampi was digested with 0.125% trypsin for 15 min, and then separated cells were prepared and fixed in ice-cold 70% ethanol at 4°C for 24 h. The fixed cells were centrifuged and washed twice in PBS. The cells were then incubated in 0.5 ml of the solution containing 200 mg/ml propidium iodide, 1% Triton, 0.9% NaCl, 50 mg/ml RNAse at 4°C for 30 min in the absence of light. Stained cells were analyzed using a FACScan Flow Cytometer. The main parameter is percentage of apoptotic cells at sub-G1 DNA peak to the total analyzed cells, which is called the apoptosis ratio.
Histopathological Observation
At the end of behavioral test, the rats deeply anaesthetized with chloral hydrate (350 mg/kg, i.p.) were perfused transcardially with 100 ml of saline followed by 400 ml of 4% paraformaldehyde. The brains were removed and paraffin-embedded, and 5-μm-thick coronal sections were cut. The sections with the dorsal hippocampus were selected at the same layers from each rat, stained with hematoxylin and eosin and evaluated by an examiner blinded to experimental conditions. Neurons with round or oval-shaped nuclei and without shrinkage or edema were counted. The number of neurons along 1 mm linear length in the middle CA1 of the hippocampus was counted, and the average value from three adjacent sections was used for each animal.
Immunohistochemical Analysis
The brain sections were thoroughly washed with 0.02 M phosphate-buffered saline (PBS, pH 7.4) for 15 min. To block any endogenous peroxidase activity, the brain sections were incubated with 0.3% hydrogen peroxide for 5 min and transferred to normal goat serum (1:20) for 90 min at room temperature, followed by overnight incubation with polyclonal rabbit-derived primary antibodies to Bax and Bcl-2, respectively (1:500 dilutions in PBS; Santa Cruz), at 4°C. After incubation with a biotinylated goat anti-rabbit secondary antibody (1:200 dilutions in PBS; Vector) for 90 min at room temperature, the sections were labeled with an avidin-biotinylated peroxidase complex (1:50 dilutions in PBS; Vector), followed by staining with 3,3′-diaminobenzidine in PBS for 5 min. The slides were observed under a microscope and photographed. A negative control was produced by PBS instead of the primary antibody. The number of positive cells was determined by counting the number of neurons possessing brown staining cytoplasm along 1 mm linear length of the CA1 hippocampus, and the average value from adjacent three sections was used for each animal. The ratio of the number of Bcl-2 positive cells over the number of Bax positive cells was expressed as Bcl/Bax ratio.
Statistical Analysis
All data were presented as mean ± SEM. Statistical significance was assessed with one-way analysis of variance (ANOVA) followed by a post hoc (Bonferroni) test for multiple group comparison. Differences with P value less than 0.05 were considered statistically significant.
Results
Histological Changes and Apoptosis after VD
To investigate the histological change after VD, different time points after 4-VO for three times were chosen. As shown in Fig. 1, pyramidal neurons in the CA1 region of hippocampus in the sham-operated group lined up in order with round and full nuclei and clear nucleolus. VD provoked many histological changes in the CA1 hippocampus such as disappearance of neurons, shrunken neurons with darkly stained condensed nuclei and the disordered and irregular pyramidal layer in the arrangement. There was no significant difference in the number of pyramidal neurons between the VD 8 h group and the sham-operated group. At 24 h after VD, Pyramidal neurons started to disappear together with infiltration of the reactive astrocytes in the CA1 region, but the effect was not statistically significant. Massive infiltration of the astrocytes and disappearance of neurons were observed and the number of pyramidal neurons was markedly decreased at 72 h after VD compared with the sham-operated group. Furthermore, the number of pyramidal neurons was significantly reduced at 168 and 720 h after VD and a tendency of gradually decreased number of pyramidal neurons was observed over 720 h after VD.

Histological changes in the CA1 region of hippocampus at 8, 24, 72, 168 and 720 h after VD. A Representative images stained with hematoxylin and eosin from sham (a) and different time points (b, 8h; c, 24h; d, 72h; e, 168h; f, 720h) after VD. Magnification = 400. B The number of pyramidal neurons in the CA1 hippocampus at different time points after VD. The number of neurons along 1 mm linear length in the middle CA1 of the hippocampus was counted. Values are means ± SE. * P < 0.05, ** P < 0.01 vs. sham, n = 5 in each group
Apoptosis after VD was further examined by flow cytometric analysis. The apoptosis ratio in the hippocampus is shown in Fig. 2A. The apoptosis ratio started to be significantly increased at 8 h after VD when compared to that of the sham group. In addition, the apoptosis ratio was gradually increased over 720 h after VD.

A Apoptosis ratio in the hippocampus at 8, 24, 72, 168 and 720 h after VD. Apoptosis was detected using flow cytometry. Apoptosis ratio was expressed as the percentage of the apoptotic cells. B H2S content in plasma at 8, 24, 72, 168 and 720 h after VD. C Correlation of apoptosis ratio in the hippocampus and H2S content in plasma. Values are means ± SE. * P < 0.05, ** P < 0.01 vs. sham, n = 5 in each group
H2S Content in Plasma and Correlation with Apoptosis Ratio in the Hippocampus after VD
To determine the change of H2S after VD, the endogenous level of H2S in plasma was assayed at different time points after VD. It was found that H2S content in plasma was markedly decreased at all of the time points after VD compared with sham rats (Fig. 2B). There was a tendency that the H2S content was gradually decreased over 720 h after VD. Furthermore, a good negative correlation between apoptosis ratio in the hippocampus and H2S content in plasma after VD was also clearly found (Fig. 2C). These findings indicate that H2S may produce a protective effect on the neural injury induced by VD.
H2S Prevents the Injury of Neurons Induced by VD
To further confirm the protective effect of H2S on VD injury, NaHS, an H2S donor or HA, an inhibitor of CBS was injected intraperitoneally 15 min before VD. Hematoxylin and eosin staining was used to show the VD-induced neuronal injury at 720 h after VD. As shown in Fig. 3A, B, VD significantly diminished the number of pyramidal neurons in the CA1 region of hippocampus compared with the sham group. Treatment with NaHS efficiently reversed these VD-induced pathological alterations with increased number of pyramidal neurons in the hippocampal CA1 region. HA treatment had a tendency to exacerbate VD-induced injury with decreased number of pyramidal neurons as compared with VD group, but the difference is not statistically significant.

A Representative images stained with hematoxylin and eosin from sham (a), VD (b), NaHS treatment (c) and HA treatment (d) groups at 720 h after VD. Magnification = 400. B The number of pyramidal neurons in the CA1 hippocampus from sham (a), VD (b), NaHS treatment (c) and HA treatment (d) groups at 720 h after VD. C Morris water maze test shows the escape latency of each group in last day of four successive days. Values are means ± SE. * P < 0.05, ** P < 0.01, n = 6 in each group
To study the effect of H2S on learning and memory after VD, a water maze was used to determine the change in learning and memory function. As shown in Fig. 3C, VD did significantly increase escape latency as compared with sham group. When treated with NaHS 15 min before VD, escape latency was markedly blunted. In contrast, treatment with HA significantly increased escape latency. These data suggest that H2S can protect the neuron against the injury induced by VD and improve learning and memory function.
The Protective Effects of H2S Involve Bcl-2 and Bax
Bcl-2, an anti-apoptotic protein, prevents the release of cytochrome c from mitochondria, whereas Bax, a pro-apoptotic protein, promotes its release. During apoptosis, Bax translocates to the outer mitochondrial membrane to induce mitochondrial membrane permeabilization. This process is blocked by the Bcl-2 protein. Therefore, the effects of NaHS on the expression of Bcl-2 and Bax in the hippocampal CA1 region were also examined. Immunohistological analysis shows that VD significantly decreased the Bcl-2 expression and increased the Bax protein expression (Fig. 4A, B). The ratio of Bcl-2 over Bax was decreased by 43% in VD rats compared with sham rats (Fig. 4C). This effect was markedly attenuated by NaHS treatment, and exaggerated by HA treatment (Fig. 4C). These data suggest that H2S prevents VD-induced injury through increasing the ratio of Bcl-2 over Bax.

A Representative images with immunohistochemical staining showing Bcl-2 expression in the CA1 region of hippocampus from sham (a), VD (b), NaHS treatment (c) and HA treatment (d) groups at 720 h after VD. Cells with brown staining cytoplasm is Bcl-2- positive. Magnification = 400. B Representative images with immunohistochemical staining showing Bax expression in the CA1 region of hippocampus from sham (a), VD (b), NaHS treatment (c) and HA treatment (d) groups at 720 h after VD. Cells with brown staining cytoplasm are Bax-positive. Magnification = 400. C Bcl/Bax ratio from sham (a), VD (b), NaHS treatment (c) and HA treatment (d) groups at 720 h after VD. Bcl/Bax ratio was represented by the ratio of Bcl-2 positive cells over Bax-positive cells in 1 mm linear length in the CA1 region of hippocampus. Values are means ± SE. * P < 0.05, ** P < 0.01, n = 6 in each group
Discussions
There are several new findings in the present study. First, we demonstrated that VD induced by 4-VO produced gradually severe neuronal injury with decreased number of neurons and increased apoptosis ratio in the hippocampus over 720 h after VD. Secondly, VD gradually decreased the H2S level in plasma over 720 h after VD, which negatively correlated with the apoptosis ratio in the hippocampus after VD. In addition, NaHS treatment significantly attenuated neuronal injury and improved neural functional performance in the Morris water maze, whereas HA exaggerated the neuronal injury and exacerbated learning and memory. Finally, NaHS treatment markedly improved the rate of Bcl-2 over Bax with increased Bcl-2 expression and decreased Bax expression, but HA reduced the ratio of Bcl-2 over Bax. Our findings suggest H2S may have potential therapeutic value for VD.
Vascular dementia remains the second most common disease among age-related dementia [31] and is usually thought to be caused by various cerebrovascular injury [32]. Many studies have demonstrated that cerebral ischemia can produce cognitive deficit and neuronal damage [33]. According to clinical observation, the patients of VD are generally accompanied with cerebral ischemia and cognitive deficit [32]. The hippocampus is a key vulnerable area to cerebral ischemia. Therefore, we selected the animal model of cerebral ischemia induced by 4-VO to mimic the VD and observed the change in the hippocampus. In the present study, the VD group showed gradual injury with decreased number of neurons and increased rate of apoptosis in the hippocampus over time after VD. Neuronal loss in the CA1 area of the hippocampus as well as some glial proliferation has also been shown in VD induced by permanent bilateral common carotid artery occlusion [34]. The hippocampus plays a major role in learning and memory, and is known to be the basis for the improvement of memory performance, which relies on the integrity of the hippocampus [35]. It is possible that injury in the hippocampus and cortex further suppresses the induction of long-term potentiation, which exacerbates loss of learning and memory. In the current study, loss of learning and memory has been observed in rats after VD.
H2S possesses a number of signaling actions, which are likely to attenuate the pathological aspects of ischemia reperfusion injury. Hu et al. [36] recently reported that H2S protects against LPS-induced inflammation in both microglia and astrocytes by significantly attenuating LPS-stimulated nitric oxide and TNF-a secretion in glial cells in a concentration-dependent manner. It has also become evident that H2S is as a potent antioxidant, and under chronic conditions, can upregulate antioxidant defenses [18, 37, 38]. These beneficial effects of H2S could contribute to protection against VD injury produced by cerebral ischemia reperfusion. A recent study has revealed that in the cardioprotective effects of H2S in myocardial ischemia reperfusion injury, H2S dose-dependently attenuates myocardial infarct size and preserves postischemic left ventricular function [39]. In addition, H2S preperfusion protects rat lung from ischemia reperfusion injury through reducing MDA and potentiating SOD [22]. Similarly, Jha et al. [21] have demonstrated that H2S can protect the murine liver against ischemia reperfusion injury through an upregulation of intracellular antioxidant pathways. In our study, we found gradually decreased H2S level in plasma over 720 h after VD. This is consistent with observation made in rats with myocardial ischemia [40]. Impaired activity [41] and reduced protein expression [42] of CBS and CSE may contribute to the decrease of plasma H2S. In addition, H2S level in plasma exhibited significantly negative correlation with apoptosis in the hippocampus in the present study. It appeared that H2S could attenuate VD injury induced by cerebral ischemia reperfusion. Similar to results in the studies of heart, lung and liver, our further study confirmed that NaHS treatment could reduce neuronal injury and improve neural functional performance, whereas HA treatment exaggerated the neuronal injury and exacerbated learning and memory. Taken together, it appears that H2S can protect a variety of organs and possesses all of the positive effects of another gaseous signaling molecule, such as NO and CO without their adverse effects [43].
However, the biological effects of H2S in the nervous system are complex. There are some controversial results in which middle cerebral artery occlusion is shown to increase tissue H2S in the cerebral cortex, and four different inhibitors of H2S synthesis are able to reduce infarct volume according to their ability in inhibiting CBS [44]. Animal model, and concentration of H2S may account for this difference. It is thought that a low concentration of H2S could offer a protective effect on the cells while higher H2S exposure tends to be cytotoxic to the cells [45]. In the present study, 14 μmol/kg of NaHS was intraperitoneally administered and it was confirmed that the dose of NaHS conferred a protective effect on neuronal injury induced by VD. Interestingly, it was also reported that H2S can be rapidly absorbed in a kind of sulfur store in a form of bound sulfane sulfur and then gradually released upon stimulation [46, 47]. Therefore, a low concentration of H2S may be offered continually into the body and this may also explain the persistent protective effects of H2S despite its short half-life.
Although the mechanisms of VD injury are not clear, inflammation [48], oxidative stress [34], apoptosis [49], vascular factors [50–52] and diabetes [52] are thought to play important roles in VD injury.
Apoptosis, delayed cell death, plays a significant role in the pathophysiology of cerebral ischemia reperfusion injury [53] and also in VD injury [24]. Masumura et al. [49] reported that the apoptosis with increased TUNEL-positive cells also resulted in white matter injury after VD in rats. Similarly, in the present study, VD rats showed gradual increase in the apoptosis ratio in the hippocampus with time after VD. Previously, in vitro studies demonstrated that H2S abolished β-phenylethyl isothiocyanate-induced apoptotic cell death in the human adenocarcinoma cell line HCT116 [54] and hydrogen sulfide prevents apoptosis of human polymorphonuclear cells via inhibition of p38 and caspase 3 [25], suggesting its potential to be a potent inhibitor of apoptosis aside from its anti-inflammation, anti-oxidative stress and vasorelaxation effects. In an in vivo study, hydrogen sulfide has previously been shown to attenuate myocardial ischemia-reperfusion injury by preservation of mitochondrial function and inhibiting apoptosis [39]. Jha et al. [21] have very recently demonstrated that H2S attenuates hepatic ischemia reperfusion injury through an anti-apoptotic signaling pathway. In the current study, H2S content in plasma significantly correlated with apoptosis in the hippocampus, suggesting its protective effects against apoptosis. Bcl-2, an anti-apoptotic protein, prevents the release of cytochrome c from mitochondria, whereas Bax, a pro-apoptotic protein, promotes its release. During apoptosis, Bax translocates to the outer mitochondrial membrane to induce mitochondrial membrane permeabilization. This process is blocked by Bcl-2 protein. Apoptosis manifests in two major execution programs downstream of the death signal: the caspase pathway and organelle dysfunction, of which mitochondrial dysfunction is the best characterized [55]. The ratio of pro-apoptotic (Bax) to anti-apoptotic (Bcl-2) members is a major checkpoint in the common portion of this apoptotic pathway and plays a pivotal role in deciding whether a cell will live or die [56]. To further confirm the anti-apoptotic effect of H2S, we determined the Bcl-2 and Bax expression in the hippocampus. The results showed that NaHS-treated rats had a greater ratio of Bcl-2 over Bax with increased Bcl-2 expression and decreased Bax expression in the hippocampus. Taken together, it suggests that H2S could protect the brain against VD injury induced by cerebral ischemia reperfusion through inhibiting the apoptosis in the hippocampus. Further studies are required to determine other mechanisms of the protective effect of H2S on VD injury to further understand the role of H2S under pathological conditions.
In summary, our findings demonstrate that H2S significantly attenuates neuronal injury induced by VD via suppressing the apoptosis in the hippocampus. These results suggest H2S may provide a potential value for treating VD injury.
References
Emery VO, Gillie EX, Smith JA (2005) Noninfarct vascular dementia and Alzheimer dementia spectrum. J Neurol Sci 229–230:27–36. doi:10.1016/j.jns.2004.11.016
Roman GC (2002) Vascular dementia may be the most common form of dementia in the elderly. J Neurol Sci 203–204:7–10. doi:10.1016/S0022-510X(02)00252-6
Rockwood K, Wentzel C, Hachinski V et al (2000) Prevalence and outcomes of vascular cognitive impairment. Vascular cognitive impairment investigators of the Canadian study of health and aging. Neurology 54:447–451
Roman GC (2002) Vascular dementia revisited: diagnosis, pathogenesis, treatment, and prevention. Med Clin North Am 86:477–499. doi:10.1016/S0025-7125(02)00008-1
Smith RP, Gosselin RE (1979) Hydrogen sulfide poisoning. J Occup Med 21:93–97. doi:10.1097/00043764-197902000-00008
Leslie M (2008) Medicine. Nothing rotten about hydrogen sulfide’s medical promise. Science 320:1155–1157. doi:10.1126/science.320.5880.1155
Li L, Moore PK (2008) Putative biological roles of hydrogen sulfide in health and disease: a breath of not so fresh air? Trends Pharmacol Sci 29:84–90. doi:10.1016/j.tips.2007.11.003
Abe K, Kimura H (1996) The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci 16:1066–1071
Qu K, Lee SW, Bian JS et al (2008) Hydrogen sulfide: neurochemistry and neurobiology. Neurochem Int 52:155–165. doi:10.1016/j.neuint.2007.05.016
Lowicka E, Beltowski J (2007) Hydrogen sulfide (H2S)—the third gas of interest for pharmacologists. Pharmacol Rep 59:4–24
Zhao W, Zhang J, Lu Y et al (2001) The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J 20:6008–6016. doi:10.1093/emboj/20.21.6008
Wang YF, Mainali P, Tang CS et al (2008) Effects of nitric oxide and hydrogen sulfide on the relaxation of pulmonary arteries in rats. Chin Med J (Engl) 121:420–423
Koenitzer JR, Isbell TS, Patel HD et al (2007) Hydrogen sulfide mediates vasoactivity in an O2-dependent manner. Am J Physiol Heart Circ Physiol 292:H1953–H1960. doi:10.1152/ajpheart.01193.2006
Cheng Y, Ndisang JF, Tang G et al (2004) Hydrogen sulfide-induced relaxation of resistance mesenteric artery beds of rats. Am J Physiol Heart Circ Physiol 287:H2316–H2323. doi:10.1152/ajpheart.00331.2004
Zanardo RC, Brancaleone V, Distrutti E et al (2006) Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J 20:2118–2120. doi:10.1096/fj.06-6270fje
Muzaffar S, Shukla N, Bond M et al (2008) Exogenous hydrogen sulfide inhibits superoxide formation, NOX-1 expression and Rac1 activity in human vascular smooth muscle cells. J Vasc Res 45:521–528. doi:10.1159/000129686
Muzaffar S, Jeremy JY, Sparatore A et al (2008) H(2)S-donating sildenafil (ACS6) inhibits superoxide formation and gp91(phox) expression in arterial endothelial cells: role of protein kinases A and G. Br J Pharmacol 155:984–994. doi:10.1038/bjp.2008.326
Kimura Y, Kimura H (2004) Hydrogen sulfide protects neurons from oxidative stress. FASEB J 18:1165–1167
Umemura K, Kimura H (2007) Hydrogen sulfide enhances reducing activity in neurons: neurotrophic role of H2S in the brain? Antioxid Redox Signal 9:2035–2041. doi:10.1089/ars.2007.1802
Sodha NR, Clements RT, Feng J et al (2008) The effects of therapeutic sulfide on myocardial apoptosis in response to ischemia-reperfusion injury. Eur J Cardiothorac Surg 33:906–913. doi:10.1016/j.ejcts.2008.01.047
Jha S, Calvert JW, Duranski MR et al (2008) Hydrogen sulfide attenuates hepatic ischemia-reperfusion injury: role of antioxidant and antiapoptotic signaling. Am J Physiol Heart Circ Physiol 295:H801–H806. doi:10.1152/ajpheart.00377.2008
Fu Z, Liu X, Geng B et al (2008) Hydrogen sulfide protects rat lung from ischemia-reperfusion injury. Life Sci 82:1196–1202. doi:10.1016/j.lfs.2008.04.005
Strub R (2003) Vascular dementia. South Med J 96:363–366. doi:10.1097/01.SMJ.0000063470.95541.6B
Yagami T (2006) Cerebral arachidonate cascade in dementia: Alzheimer’s disease and vascular dementia. Curr Neuropharmacol 4:87–100. doi:10.2174/157015906775203011
Rinaldi L, Gobbi G, Pambianco M et al (2006) Hydrogen sulfide prevents apoptosis of human PMN via inhibition of p38 and caspase 3. Lab Invest 86:391–397. doi:10.1038/labinvest.3700391
Hu LF, Lu M, Wu ZY et al (2009) Hydrogen sulfide inhibits rotenone-induced apoptosis via preservation of mitochondrial function. Mol Pharmacol 75:27–34. doi:10.1124/mol.108.047985
Zhu YZ, Wang ZJ, Ho P et al (2007) Hydrogen sulfide and its possible roles in myocardial ischemia in experimental rats. J Appl Physiol 102:261–268. doi:10.1152/japplphysiol.00096.2006
Reiffenstein RJ, Hulbert WC, Roth SH (1992) Toxicology of hydrogen sulfide. Annu Rev Pharmacol Toxicol 32:109–134. doi:10.1146/annurev.pa.32.040192.000545
Gilboa-Garber N (1971) Direct spectrophotometric determination of inorganic sulfide in biological materials and in other complex mixtures. Anal Biochem 43:129–133. doi:10.1016/0003-2697(71)90116-3
Morris R (1984) Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods 11:47–60. doi:10.1016/0165-0270(84)90007-4
Wu WW, Oh MM, Disterhoft JF (2002) Age-related biophysical alterations of hippocampal pyramidal neurons: implications for learning and memory. Ageing Res Rev 1:181–207. doi:10.1016/S1568-1637(01)00009-5
Knopman DS (2007) Cerebrovascular disease and dementia. Br J Radiol 80(Spec No 2):S121–S127
Zarow C, Vinters HV, Ellis WG et al (2005) Correlates of hippocampal neuron number in Alzheimer’s disease and ischemic vascular dementia. Ann Neurol 57:896–903. doi:10.1002/ana.20503
Liu C, Wu J, Gu J et al (2007) Baicalein improves cognitive deficits induced by chronic cerebral hypoperfusion in rats. Pharmacol Biochem Behav 86:423–430. doi:10.1016/j.pbb.2006.11.005
Olton DS, Papas BC (1979) Spatial memory and hippocampal function. Neuropsychologia 17:669–682. doi:10.1016/0028-3932(79)90042-3
Hu LF, Wong PT, Moore PK et al (2007) Hydrogen sulfide attenuates lipopolysaccharide-induced inflammation by inhibition of p38 mitogen-activated protein kinase in microglia. J Neurochem 100:1121–1128. doi:10.1111/j.1471-4159.2006.04283.x
Wei HL, Zhang CY, Jin HF et al (2008) Hydrogen sulfide regulates lung tissue-oxidized glutathione and total antioxidant capacity in hypoxic pulmonary hypertensive rats. Acta Pharmacol Sin 29:670–679. doi:10.1111/j.1745-7254.2008.00796.x
Yonezawa D, Sekiguchi F, Miyamoto M et al (2007) A protective role of hydrogen sulfide against oxidative stress in rat gastric mucosal epithelium. Toxicology 241:11–18. doi:10.1016/j.tox.2007.07.020
Elrod JW, Calvert JW, Morrison J et al (2007) Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci USA 104:15560–15565. doi:10.1073/pnas.0705891104
Zhu XY, Yan XH, Chen SJ (2008) H2S protects myocardium against ischemia/reperfusion injury and its effect on c-Fos protein expression in rats. Sheng Li Xue Bao 60:221–227
Prathapasinghe GA, Siow YL, Xu Z et al (2008) Inhibition of cystathionine-beta-synthase activity during renal ischemia-reperfusion: role of pH and nitric oxide. Am J Physiol Renal Physiol 295:F912–F922. doi:10.1152/ajprenal.00040.2008
Sen U, Vacek TP, Hughes WM et al (2008) Cardioprotective role of sodium thiosulfate on chronic heart failure by modulating endogenous H2S generation. Pharmacology 82:201–213. doi:10.1159/000156486
Lefer DJ (2007) A new gaseous signaling molecule emerges: cardioprotective role of hydrogen sulfide. Proc Natl Acad Sci USA 104:17907–17908. doi:10.1073/pnas.0709010104
Qu K, Chen CP, Halliwell B et al (2006) Hydrogen sulfide is a mediator of cerebral ischemic damage. Stroke 37:889–893. doi:10.1161/01.STR.0000204184.34946.41
Szabo C (2007) Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov 6:917–935. doi:10.1038/nrd2425
Ogasawara Y, Ishii K, Togawa T et al (1993) Determination of bound sulfur in serum by gas dialysis/high-performance liquid chromatography. Anal Biochem 215:73–81. doi:10.1006/abio.1993.1556
Warenycia MW, Goodwin LR, Francom DM et al (1990) Dithiothreitol liberates non-acid labile sulfide from brain tissue of H2S-poisoned animals. Arch Toxicol 64:650–655. doi:10.1007/BF01974693
Malaguarnera L, Motta M, Di Rosa M et al (2006) Interleukin-18 and transforming growth factor-beta 1 plasma levels in Alzheimer’s disease and vascular dementia. Neuropathology 26:307–312. doi:10.1111/j.1440-1789.2006.00701.x
Masumura M, Hata R, Nagai Y et al (2001) Oligodendroglial cell death with DNA fragmentation in the white matter under chronic cerebral hypoperfusion: comparison between normotensive and spontaneously hypertensive rats. Neurosci Res 39:401–412. doi:10.1016/S0168-0102(01)00195-X
Brown WR, Thore CR, Moody DM et al (2005) Vascular damage after fractionated whole-brain irradiation in rats. Radiat Res 164:662–668. doi:10.1667/RR3453.1
Brown WR, Blair RM, Moody DM et al (2007) Capillary loss precedes the cognitive impairment induced by fractionated whole-brain irradiation: a potential rat model of vascular dementia. J Neurol Sci 257:67–71. doi:10.1016/j.jns.2007.01.014
Stephan BC, Brayne C (2008) Vascular factors and prevention of dementia. Int Rev Psychiatry 20:344–356. doi:10.1080/09540260802094456
Doyle KP, Simon RP, Stenzel-Poore MP (2008) Mechanisms of ischemic brain damage. Neuropharmacology 55:310–318. doi:10.1016/j.neuropharm.2008.01.005
Rose P, Moore PK, Ming SH et al (2005) Hydrogen sulfide protects colon cancer cells from chemopreventative agent beta-phenylethyl isothiocyanate induced apoptosis. World J Gastroenterol 11:3990–3997
Green DR, Reed JC (1998) Mitochondria and apoptosis. Science 281:1309–1312. doi:10.1126/science.281.5381.1309
Gross A, McDonnell JM, Korsmeyer SJ (1999) BCL-2 family members and the mitochondria in apoptosis. Genes Dev 13:1899–1911. doi:10.1101/gad.13.15.1899
Acknowledgments
The study was supported by grants from Excellent Youth Foundation of Yunnan Scientific Committee (2006WFX04) and Foundation of Yunnan Educational Committee (06Y089C).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhang, LM., Jiang, CX. & Liu, DW. Hydrogen Sulfide Attenuates Neuronal Injury Induced by Vascular Dementia Via Inhibiting Apoptosis in Rats. Neurochem Res 34, 1984–1992 (2009). https://doi.org/10.1007/s11064-009-0006-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-009-0006-9