
Abstract
Disulfiram, a generic alcohol aversion drug, has promising preclinical activity against glioblastoma (GBM). This phase I study aims to evaluate its safety, maximum tolerated dose (MTD), pharmacodynamic effect, and preliminary efficacy when combined with adjuvant temozolomide in GBM patients after standard chemoradiotherapy. Patients received disulfiram 500–1000 mg once daily, in combination with 150–200 mg/m2 temozolomide. A modified 3 + 3 dose-escalation design was used to determine the MTD. The pharmacodynamic effect of proteasome inhibition was assessed using fluorometric 20S proteasome assay on peripheral blood cells. The MTD was determined based on the dose-limiting toxicities (DLTs) within the first month of therapy. Twelve patients were enrolled to two dose levels: 500 and 1000 mg. Two DLTs of grade 3 delirium occurred after 15 days of administration at 1000 mg per day. Other possible grade 2–3 DSF-related toxicities included fatigue, ataxia, dizziness, and peripheral neuropathy. The toxicities were self-limiting or resolved after discontinuing DSF. The MTD was determined to be 500 mg per day. Limited proteasome inhibition was observed at week 4 and showed an increased trend with escalated disulfiram. Median progression-free survival with 500 mg of DSF was 5.4 months from the start of disulfiram and 8.1 months from the start of chemoradiotherapy. Disulfiram can be safely combined with temozolomide but can cause reversible neurological toxicities. The MTD of disulfiram with adjuvant temozolomide appears to produce limited proteasome inhibition on peripheral blood cells.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Glioblastoma multiforme (GBM, World Health Organization/WHO grade IV) is the most common malignant primary brain tumor in adults and one of the most devastating cancers [1]. Between 2004 and 2007, there were 37,690 patients newly diagnosed with GBM, with an estimated incidence rate of three cases per 100,000 people in the United States [2]. The current standard of care for GBM includes maximal safe resection, radiation therapy (RT) with concurrent temozolomide (TMZ), followed by adjuvant TMZ. However, despite such multi-modality therapy, the median survival is approximately 14 months, with a 5-year survival of less than 10 % [3]. Thus, novel therapeutic approaches are desperately needed for this challenging disease.
Besides the aggressive and heterogeneous biology of GBM, economical factors have also presented challenges for advancing treatments. The expense of developing a new oncology drug is approximately $1 billion and rising [4], which translates to very expensive cost per patient, typically in the range of $5000–$10,000 per month, or $50,000–$100,000 per course [5]. GBM is also considered a rare disease by the FDA and an orphan disease by the pharmaceutical companies, thus further limiting financial incentive for drug development [6]. Therefore, there has been growing interest to repurpose previously approved non-cancer drugs as potential anti-cancer treatments, which offers advantages of shorter development time, lower research cost, and cheaper drug price [5]. One of the most promising drugs to repurpose for treating GBM is disulfiram (DSF) [7, 8]. DSF is a FDA-approved oral medication that has been used for treating alcoholism since 1951. It inhibits aldehyde dehydrogenase (ALDH), which leads to accumulation of acetaldehyde in the blood after ingestion of alcohol. It has well-known safety profile for up to 3000 mg per day in the absence of alcohol consumption, well-established pharmacokinetics, and ability to readily cross the blood–brain barrier (BBB) [9, 10]. Multiple preclinical in vitro studies have demonstrated its promising activity against glioma stem-like cells (GSCs) [11–13]. GSCs represent a subset of GBM tumor cells that have been shown to be more resistant to RT [14]. In an unbiased high-throughput screening of 2000 compounds using GSCs from multiple GBM patients, DSF was identified to have one of the most potent anti-tumor activity and appeared relatively non-toxic to human neural stem cells [11]. DSF also has synergistic activity when combined with TMZ and is highly effective against TMZ-resistance cells [13]. Multiple studies have suggested the mechanism of DSF’s anti-cancer property is due to inhibition of 20S proteasome activity, which is dependent on complex formation with copper (Cu) [11, 15, 16]. In vivo studies have also confirmed that DSF and Cu have synergistic effects with TMZ and can improve survival of mice with orthotopic GBM tumors [17, 18].
This is a report of the first clinical evaluation of DSF to treat GBM based on an open-label, phase I, dose-escalation pharmacodynamic study for newly diagnosed GBM patients after standard chemoradiotherapy. The primary objective of this study was to determine the safety and the maximum tolerated dose (MTD) of DSF in combination with adjuvant TMZ. Secondary objectives were to evaluate the pharmacodynamic effect of proteasome inhibition and to assess preliminary anti-tumor activity.
Materials and methods
Patient selection
Eligible patients were required to be age ≥18 years with newly diagnosed GBM and were eligible to receive adjuvant TMZ after completion of standard RT with concurrent TMZ. Additional eligibilities included: Zubrod performance score of 0–2, abstinence from alcohol consumption, adequate bone marrow, liver, and renal functions (absolute neutrophil count ≥1500 per cubic millimeter; platelet ≥100,000 per cubic millimeter; total bilirubin ≤2× upper limit of normal [ULN]; liver-function values <3× ULN; and creatinine clearance >60 mL/min). Patients were excluded if they had idiopathic seizure disorder, psychosis, or schizophrenia. Medications with significant cytochromes P450 enzyme activity and interaction with DSF were not permitted. All patients gave written informed consent. This study received Institutional Review Board approval and was conducted in accordance with the Declaration of Helsinki and good clinical practice. The use of DSF for this study was granted exemption status by the FDA. This trial is registered with ClinicalTrials.gov, NCT01907165.
Study design
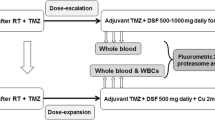
This was an open-label, single-arm, single-institution, dose-escalation study of DSF in combination with adjuvant TMZ in newly diagnosed GBM patients after standard chemoradiotherapy. Adjuvant TMZ was administered for six cycles as per routine clinical care, at a dosage of 150–200 mg/m2 daily on days 1–5 of every 28 day cycle. DSF was administered orally once a day during adjuvant TMZ (Fig. 1). The first dose level was initiated at 500 mg per day, the upper limit of current FDA-recommended dose for alcohol abstinence. Six patients per dose level were planned to allow assessment of pharmacodynamic effects and tolerability. Dose-escalation criteria of this study was modified from the traditional 3 + 3 design to account for both dose-limiting toxicity (DLT) and proteasome inhibition (the hypothesized mechanism of action based on preclinical data). Dose escalation was permitted only if no more than one patient (<33 %) in the current dose level experienced DLT and at least two patients had <90 % proteasome inhibition. The rationale was if significant target-specific action had been achieved, further dose-escalation would not be necessary. Six patients were allowed to enroll per cohort to allow an accurate estimate of proteasome inhibition and because of the well-known safety profile of DSF based on extensive clinical use since 1951. Patients were instructed not to take disulfiram within 1 h of TMZ administration and to take it with food to improve absorption.

Schema. Abbreviations GBM, glioblastoma; RT, radiation therapy; TMZ, temozolomide
Safety and response evaluation
Before study entry, all patients underwent physical examination, standard laboratory studies, and MRI of the brain. Patients were seen monthly before the start of each cycle of TMZ. Liver function tests including alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were evaluated monthly, and MRIs of the brain were performed every 2 months or as clinically indicated. Tumor response or progression was evaluated using the response assessment in neuro-oncology (RANO) criteria [19]. A DLT was defined as the occurrence of any of the following toxicities that were possibly, probably, or definitely related to DSF within the first 28 days of starting DSF: grade 4 neutropenia >21 days, grade 4 thrombocytopenia >28 days, grade 3 or higher non-hematological toxicities (excluding grade 3 fatigue and myalgias, grade 3 nausea, or laboratory abnormalities that resolved prior to the next cycle of TMZ). The MTD was defined as the dose level immediately below the dose at which at least two patients experienced DLTs. Patients were evaluated for adverse events from the first dose of study treatment until a 30-day follow up after the conclusion of treatment or death. Toxicity was graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) version 4.0.
Pharmacodynamic analysis
Preclinical data indicate that the anti-tumor mechanism of DSF is through inhibition of chymotrypsin-like 20S proteasome activity [11, 15, 16]; this study was designed to evaluate its pharmacodynamic effect on proteasome activity in patients. Since repeated biopsies of GBM to directly assess proteasome inhibition would be difficult, peripheral blood cells were used as surrogate markers. Blood samples were collected before the start of DSF, after 2 weeks, and after 4 weeks/before the second cycle of TMZ (Fig. 1). Measurements of chymotrypsin-like 20S proteasome activities were performed with fluorometric 20S proteasome assay as previously described using whole blood lysate and fluorogenic substrate Suc-LLVY-AMC [20], which has been tested extensively in clinical trials of proteasome inhibitors for multiple myeloma [21, 22].
Statistical analysis
All continuous measurements were summarized by mean (or median for non-normal data), SD, minimum, maximum. Categorical data were summarized by frequency counts and percentages. Time to event was calculated with two methods: from the start of DSF and from the start of RT to allow comparison to historical data from randomized trials. Progression-free survival (PFS) and overall survival (OS) were estimated with Kaplan–Meier method. Significance was defined as a P value ≤0.05. Statistical analyses were performed with the Statistical Package for Social Sciences, version 17.0 (IBM SPSS Statistics, Chicago, IL, USA). Proteasome activity was normalized to baseline activity and analyzed by Kruskal–Wallis test followed by Dunn’s procedure for multiple pair-wise comparisons. All statistical tests were two sided.
Results
Patient characteristics
A total of 14 supratentorial primary GBM patients were enrolled from October 2013 to April 2015. Two patients withdrew consent before the start of therapy, leaving 12 evaluable patients: seven patients at 500 mg per day and five patients at 1000 mg per day. The first patient enrolled to the second dose level was instructed to take 500 mg per day by mistake and was therefore evaluated as part of the first dose level. Only five patients were enrolled to the second dose level due to DLTs. The baseline patient characteristics are summarized in Table 1. The median age was 57 (range 20–75). Two patients (17 %) had biopsy; 7 (58 %) had subtotal resection; 3 (25 %) had gross-total resection. All patients completed standard RT of 60 Gy with concurrent TMZ at 75 mg/m2. Four patients (33 %) had O6-methylguanine-DNA methyltransferase (MGMT) promoter methylation.
Safety and maximum tolerated dose
None of the seven patients in the 500 mg cohort had DLTs, though two of them (29 %) had possible grade 3 DSF-related toxicities of delirium and peripheral motor neuropathy after the first month (after 55 and 80 days, respectively) that prompted discontinuation of the drug. The other patients were removed from the study due to unrelated toxicities or tumor progression. Two of the five patients in the 1000 mg cohort had DLTs. Both were grade 3 delirium and occurred after 15 days of DSF. The study was halted for further enrollment. As per protocol design, the MTD of DSF in combination with adjuvant TMZ was determined to be 500 mg per day. Two other patients in the 1000 mg cohort discontinued DSF due to grade 2–3 ataxia (after 15 and 33 days, respectively) that did not improve with a brief trial of dose reduction. The duration of DSF and the reasons for discontinuation are summarized in Table 2. The treatment-related toxicities were mostly neurological and included fatigue, delirium, ataxia, dizziness, and peripheral motor/sensory neuropathy. The toxicities were self-limiting or resolved within 30 days after discontinuing DSF. The grade 2–3 toxicities that were possibly/probably related to DSF are summarized in Table 3. Grade 2–3 adverse events that were assessed to be unlikely related to DSF are provided in Supplementary Table 1S. They were mostly hematological toxicities related to TMZ (lymphopenia, anemia, or thrombocytopenia) or symptoms related to tumor progression (ataxia, urinary incontinence, or seizure). There were no grade 4–5 adverse events.
Proteasome inhibition
All 12 patients were analyzed for chymotrypsin-like 20S proteasome activity in whole blood lysates. Three of five patients in the 1000 mg cohort completed only 2 weeks of DSF and were evaluated only for their week 2 proteasome activities. At week 2, there was little to no proteasome inhibition detected with either dosage compared to that of pretreatment baseline but rather a trend toward slightly increased activity (Fig. 2). At week 4, there was a trend toward inhibition of proteasome activity compared to that of baseline, where 500 mg of DSF was associated with a mean decrease of 5 % (SD 12 %) in proteasome activity and 1000 mg of DSF with a mean decrease of 11 % (SD 23 %) (Fig. 2).

The effect of disulfiram (DSF) dosage and duration on proteasome activity. Chymotrypsin-like 20S proteasome activities of patients taking 500 mg per day (solid columns) and 1000 mg per day (striped columns) were measured at baseline, after 2 weeks, and after 4 weeks of treatment. Proteasome activities were measured using fluorogenic substrate Suc-LLVY-AMC in whole blood lysates and normalized to pretreatment baseline value. Columns and error bars represent the mean and SD, respectively. P values were determined using the Kruskal–Wallis test and compared to the corresponding baseline activity
Preliminary efficacy
In this study, the value of radiographic response is limited as the relative contributions of RT and TMZ cannot be distinguished. Before study registration, the majority of the enrolled patients (58 %) had stable disease, four patients (33 %) had possible pseudoprogression, and one patient (patient #2, Table 2) had progression with leptomeningeal dissemination. At 2 months from starting DSF, eight patients (67 %) had either stable disease or partial response. At the time of analysis, the median follow-up time is 6.9 months (range: 2.7–24.3) with at least 6 months of follow-up for all living patients. Six of seven patients from the 500 mg cohort had progressed, and 3 of 5 patients from the 1000 mg cohort had progressed. Four patients have died, all from the 500 mg cohort. When measured from the start of DSF, the median PFS and OS for the 500 mg cohort were 5.4 months (95 % CI 0–14.8) and 12.1 months (95 % CI 2.2–22.0), respectively; 3.1 months (95 % CI 0–6.7) and not reached (NR) for the 1000 mg cohort, respectively. If calculated from the start of RT, the median PFS and OS for the 500 mg cohort were 8.1 months (0–17.3) and 14.7 months (95 % CI 4.9–24.5), respectively; 5.4 months (2.5–8.4) and NR for the 1000 mg cohort, respectively.
Discussion
This phase I, dose-escalation study evaluated oral DSF in combination with adjuvant TMZ for newly diagnosed GBM patients who had completed RT and concurrent TMZ. To our knowledge, this clinical trial is the first to test DSF as an anti-cancer treatment for GBM. Oral once daily administration of DSF in combination with TMZ demonstrated an acceptable safety profile in patients with GBM. The DLTs occurred at 1000 mg per day, and the MTD of DSF in combination with adjuvant TMZ was determined to be 500 mg per day. Most common limiting toxicities were reversible neurological toxicities and included delirium, ataxia, and neuropathy. The pharmacodynamic effect of DSF on proteasome inhibition appeared to be limited, at least when using peripheral blood cells as surrogates and without concurrent Cu administration.
DSF-related toxicities in this study are mostly neurological and are consistent with other phase I studies. Although neurological toxicities can be difficult to distinguish from tumor effect, the DSF-related neurological toxicities occurred in the absence of radiological progression and showed dramatic increase in severity and shorter time of onset with increasing DSF dose. Schweizer et al. conducted a phase I study of 250–500 mg of DSF alone in 19 prostate cancer patients, and they also observed ataxia, dizziness, neuropathy, and fatigue [23]. Stewart et al. conducted a phase I study that administered a single dose of oral DSF prior to cisplatin every 3 weeks, and dose-limiting reversible confusion occurred at 3000 mg/m2 (approximately 4800 mg) [24]. High doses of DSF may inhibit cerebrospinal dopamine B-hydroxylase [25], and people with very low activity of dopamine B-hydroxylase may be more prone to transient psychosis with such inhibition [26]. Our data suggest that continuous daily administration, intracranial GBM, and additive effect of TMZ may further increase susceptibility to such neurological effect. The neurological toxicity profile of DSF provides indirect evidence that DSF crosses the BBB. Nechushtan et al. have recently reported their preliminary results of a randomized phase IIb study of cisplatin/vinorelbine with and without DSF for metastatic non-small cell lung cancer. They did not report any significant neurological toxicity except fatigue, but the dosing of DSF was 40 mg three times per day (TID). Given the half-life of DSF is approximately 7 h [27], TID administration may improve its efficacy as anti-cancer therapy while reducing its toxicity. Further investigation of the TID dosing schedule is warranted, which our group is currently developing.
Although previous preclinical studies have suggested proteasome inhibition as the mechanism of DSF’s anti-cancer property [11, 15, 16], this study represents the first attempt to validate it in patients using peripheral blood cells as surrogate markers. At the MTD of 500 mg per day, only minimal proteasome inhibition was observed after 4 weeks, a mean reduction of approximately 5 %. However, there appeared to be a trend toward dose–response with escalating DSF, with doubling of proteasome inhibition at 1000 mg per day (Fig. 2). The limited proteasome inhibition may be due to lack of concurrent Cu administration [11, 12] or possible differential effect of DSF on normal tissue versus tumor [11]. Furthermore, other mechanisms may also be responsible for the anti-tumor property of DSF, such as ALDH inhibition [28, 29] or generation of intracellular reactive oxygen species (ROS) [12]. To address these questions, our group has developed follow-up studies to administer DSF concurrently with Cu and to directly assess proteasome inhibition in GBM tumors by administering DSF before surgery.
Given this is a small phase I study, secondary endpoints such as PFS only serve to provide preliminary signal for possible efficacy of DSF and should be interpreted with caution. During the enrollment period, there were also multiple competing phase III vaccination trials at our institution that preferentially selected for patients who had greater extent of resection or favorable response to chemoradiotherapy. Therefore, the patients from this trial were negatively selected as they did not qualify for the more stringent trials. Since this is a phase I study, we also did not exclude those who had shown possible or definitive tumor progression after chemoradiotherapy. Nevertheless, the median PFS from the start of RT for the seven patients who were treated at the MTD was 8.1 months, which compares favorably to the historical data of standard RT plus TMZ from randomized studies: 6.9 months (95 % CI 5.8–8.2) in the Stupp study [3], 5.5 months (95 % CI 4.7–6.1) in RTOG 0525 [30], and 7.3 months (95 % CI 5.9–7.9) in RTOG 0825 [31]. Although small number of patients and relatively short follow-up prevent any conclusion regarding the efficacy of the regimen, these preliminary findings are encouraging and support further investigation to study DSF.
In summary, this phase I study of daily DSF in combination with adjuvant TMZ in newly diagnosed GBM patients after standard chemoradiotherapy has established the MTD as 500 mg per day. The combination has an acceptable safety profile and produces promising PFS. It can cause reversible neurological toxicities such as delirium, ataxia, and neuropathy. In the absence of concurrent Cu administration, DSF produced limited proteasome inhibition on peripheral blood cells. Given the devastating outcomes of GBM and the enormous economical advantages of repurposing DSF, our findings support continued clinical trial development of DSF. In future directions, our group is currently developing additional clinical trials to combine DSF with concurrent Cu administration, to test the TID dosing regimen of DSF, and to administer DSF before surgical resection of GBM to allow direct assessment of intratumoral drug concentration, proteasome inhibition and other potential mechanisms of action. The results of these studies will clarify the questions generated from this study and further explore the potential of repurposing DSF to treat GBM.
References
Johnson DR, O’Neill BP (2012) Glioblastoma survival in the United States before and during the temozolomide era. J Neurooncol 107:359–364. doi:10.1007/s11060-011-0749-4
CBTRUS Statistical Report (2011): primary brain and central nervous system tumors diagnosed in the United States in 2004–2007. www.cbtrus.org
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996. doi:10.1056/NEJMoa043330
DiMasi JA, Grabowski HG (2007) Economics of new oncology drug development. J Clin Oncol 25:209–216. doi:10.1200/JCO.2006.09.0803
Pantziarka P, Bouche G, Meheus L, Sukhatme V, Sukhatme VP, Vikas P (2014) The repurposing drugs in oncology (ReDO) project. Ecancermedicalscience 8:442. doi:10.3332/ecancer.2014.442
Sharma A, Jacob A, Tandon M, Kumar D (2010) Orphan drug: development trends and strategies. J Pharm Bioallied Sci 2:290–299. doi:10.4103/0975-7406.72128
Kast RE, Boockvar JA, Bruning A, Cappello F, Chang WW, Cvek B, Dou QP, Duenas-Gonzalez A, Efferth T, Focosi D, Ghaffari SH, Karpel-Massler G, Ketola K, Khoshnevisan A, Keizman D, Magne N, Marosi C, McDonald K, Munoz M, Paranjpe A, Pourgholami MH, Sardi I, Sella A, Srivenugopal KS, Tuccori M, Wang W, Wirtz CR, Halatsch ME (2013) A conceptually new treatment approach for relapsed glioblastoma: coordinated undermining of survival paths with nine repurposed drugs (CUSP9) by the International Initiative for Accelerated Improvement of Glioblastoma Care. Oncotarget 4:502–530
Triscott J, Rose Pambid M, Dunn SE (2015) Concise review: bullseye: targeting cancer stem cells to improve the treatment of gliomas by repurposing disulfiram. Stem Cells 33:1042–1046. doi:10.1002/stem.1956
Faiman MD, Dodd DE, Hanzlik RE (1978) Distribution of S35 disulfiram and metabolites in mice, and metabolism of S35 disulfiram in the dog. Res Commun Chem Pathol Pharmacol 21:543–567
Suh JJ, Pettinati HM, Kampman KM, O’Brien CP (2006) The status of disulfiram: a half of a century later. J Clin Psychopharmacol 26:290–302. doi:10.1097/01.jcp.0000222512.25649.08
Hothi P, Martins TJ, Chen L, Deleyrolle L, Yoon JG, Reynolds B, Foltz G (2012) High-throughput chemical screens identify disulfiram as an inhibitor of human glioblastoma stem cells. Oncotarget 3:1124–1136
Liu P, Brown S, Goktug T, Channathodiyil P, Kannappan V, Hugnot JP, Guichet PO, Bian X, Armesilla AL, Darling JL, Wang W (2012) Cytotoxic effect of disulfiram/copper on human glioblastoma cell lines and ALDH-positive cancer-stem-like cells. Br J Cancer 107:1488–1497. doi:10.1038/bjc.2012.442
Triscott J, Lee C, Hu K, Fotovati A, Berns R, Pambid M, Luk M, Kast RE, Kong E, Toyota E, Yip S, Toyota B, Dunn SE (2012) Disulfiram, a drug widely used to control alcoholism, suppresses the self-renewal of glioblastoma and over-rides resistance to temozolomide. Oncotarget 3:1112–1123
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444:756–760. doi:10.1038/nature05236
Chen D, Cui QC, Yang H, Dou QP (2006) Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res 66:10425–10433. doi:10.1158/0008-5472.CAN-06-2126
Cvek B, Milacic V, Taraba J, Dou QP (2008) Ni(II), Cu(II), and Zn(II) diethyldithiocarbamate complexes show various activities against the proteasome in breast cancer cells. J Med Chem 51:6256–6258. doi:10.1021/jm8007807
Lun X, Wells JC, Hao X, Zhang J, Grinshtein N, Kaplan D, Luchman A, Weiss S, Cairncross JG, Senger DS, Robbins S (2013) Disulfiram when combined with copper is an effective adjuvant therapy with TMZ for treatment of human gliomblastoma. Neuro-oncology 15(Suppl 3):iii52 [Abstract: ET-064]
Paranjpe A, Zhang R, Ali-Osman F, Bobustuc GC, Srivenugopal KS (2014) Disulfiram is a direct and potent inhibitor of human O6-methylguanine-DNA methyltransferase (MGMT) in brain tumor cells and mouse brain and markedly increases the alkylating DNA damage. Carcinogenesis 35:692–702. doi:10.1093/carcin/bgt366
Wen PY, Macdonald DR, Reardon DA, Cloughesy TF, Sorensen AG, Galanis E, Degroot J, Wick W, Gilbert MR, Lassman AB, Tsien C, Mikkelsen T, Wong ET, Chamberlain MC, Stupp R, Lamborn KR, Vogelbaum MA, van den Bent MJ, Chang SM (2010) Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol 28:1963–1972. doi:10.1200/JCO.2009.26.3541
Lightcap ES, McCormack TA, Pien CS, Chau V, Adams J, Elliott PJ (2000) Proteasome inhibition measurements: clinical application. Clin Chem 46:673–683
Moreau P, Pylypenko H, Grosicki S, Karamanesht I, Leleu X, Grishunina M, Rekhtman G, Masliak Z, Robak T, Shubina A, Arnulf B, Kropff M, Cavet J, Esseltine DL, Feng H, Girgis S, van de Velde H, Deraedt W, Harousseau JL (2011) Subcutaneous versus intravenous administration of bortezomib in patients with relapsed multiple myeloma: a randomised, phase 3, non-inferiority study. Lancet Oncol 12:431–440. doi:10.1016/S1470-2045(11)70081-X
Orlowski RZ, Stinchcombe TE, Mitchell BS, Shea TC, Baldwin AS, Stahl S, Adams J, Esseltine DL, Elliott PJ, Pien CS, Guerciolini R, Anderson JK, Depcik-Smith ND, Bhagat R, Lehman MJ, Novick SC, O’Connor OA, Soignet SL (2002) Phase I trial of the proteasome inhibitor PS-341 in patients with refractory hematologic malignancies. J Clin Oncol 20:4420–4427
Schweizer MT, Lin J, Blackford A, Bardia A, King S, Armstrong AJ, Rudek MA, Yegnasubramanian S, Carducci MA (2013) Pharmacodynamic study of disulfiram in men with non-metastatic recurrent prostate cancer. Prostate Cancer Prostatic Dis 16:357–361. doi:10.1038/pcan.2013.28
Stewart DJ, Verma S, Maroun JA (1987) Phase I study of the combination of disulfiram with cisplatin. Am J Clin Oncol 10:517–519
Nilsson GE, Tottmar O, Wahlstrom G (1987) Effects of aldehyde dehydrogenase inhibitors on hexobarbital sensitivity and neuroamine metabolism in rat brain. Brain Res 409:265–274
Major LF, Lerner P, Ballenger JC, Brown GL, Goodwin FK, Lovenberg W (1979) Dopamine-beta-hydroxylase in the cerebrospinal fluid: relationship to disulfiram-induced psychosis. Biol Psychiatry 14:337–344
Faiman MD, Jensen JC, Lacoursiere RB (1984) Elimination kinetics of disulfiram in alcoholics after single and repeated doses. Clin Pharmacol Ther 36:520–526
Choi SA, Choi JW, Wang KC, Phi JH, Lee JY, Park KD, Eum D, Park SH, Kim IH, Kim SK (2015) Disulfiram modulates stemness and metabolism of brain tumor initiating cells in atypical teratoid/rhabdoid tumors. Neuro-oncology 17:810–821. doi:10.1093/neuonc/nou305
Kim SK, Kim H, Lee DH, Kim TS, Kim T, Chung C, Koh GY, Kim H, Lim DS (2013) Reversing the intractable nature of pancreatic cancer by selectively targeting ALDH-high, therapy-resistant cancer cells. PLoS One 8:e78130. doi:10.1371/journal.pone.0078130
Gilbert MR, Wang M, Aldape KD, Stupp R, Hegi ME, Jaeckle KA, Armstrong TS, Wefel JS, Won M, Blumenthal DT, Mahajan A, Schultz CJ, Erridge S, Baumert B, Hopkins KI, Tzuk-Shina T, Brown PD, Chakravarti A, Curran WJ Jr, Mehta MP (2013) Dose-dense temozolomide for newly diagnosed glioblastoma: a randomized phase III clinical trial. J Clin Oncol 31:4085–4091. doi:10.1200/JCO.2013.49.6968
Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S, Won M, Jeraj R, Brown PD, Jaeckle KA, Schiff D, Stieber VW, Brachman DG, Werner-Wasik M, Tremont-Lukats IW, Sulman EP, Aldape KD, Curran WJ Jr, Mehta MP (2014) A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med 370:699–708. doi:10.1056/NEJMoa1308573
Acknowledgments
The authors would like to thank Dr. Eric Lightcap for his helpful discussions on proteasome activity assay. We would like to thank the Alvin J. Siteman Cancer Center at Washington University School of Medicine and Barnes-Jewish Hospital in St. Louis, MO., for the use of the Clinical Trials Office, which provided protocol development, regulatory, data management, and study coordination services. We would like to specifically acknowledge Stephanie Myles, Bethany Rensink, Sarah Marchetti, and Maria Miller of the Siteman Cancer Center for their work on this trial. The Siteman Cancer Center is supported in part by a NCI Cancer Center Support Grant #P30 CA091842. We would also like to acknowledge our courageous patients and their families for their inspiration.
Funding
Institutional fund from the Department of Radiation Oncology, Washington University School of Medicine.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
We have no conflict of interests to disclose for this work.
Additional information
Jian L. Campian and Amit D. Gujar contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Huang, J., Campian, J.L., Gujar, A.D. et al. A phase I study to repurpose disulfiram in combination with temozolomide to treat newly diagnosed glioblastoma after chemoradiotherapy. J Neurooncol 128, 259–266 (2016). https://doi.org/10.1007/s11060-016-2104-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-016-2104-2