
Abstract
Cerebral spinal fluid (CSF) from brain tumor patients contains tumor cellular and cell-free DNA (cfDNA), which provides a less-invasive and routinely accessible method to obtain tumor genomic information. In this report, we used droplet digital PCR to test mutant tumor DNA in CSF of a patient to monitor the treatment response of metastatic melanoma leptomeningeal disease (LMD). The primary melanoma was known to have a BRAF V600E mutation, and the patient was treated with whole brain radiotherapy and BRAF inhibitors. We collected 9 CSF samples over 6 months. The mutant cfDNA fraction gradually decreased from 53 % (time of diagnosis) to 0 (time of symptom alleviation) over the first 6 time points. Three months after clinical improvement, the patient returned with severe symptoms and the mutant cfDNA was again detected in CSF at high levels. The mutant DNA fraction corresponded well with the patient’s clinical response. We used whole exome sequencing to examine the mutation profiles of the LMD tumor DNA in CSF before therapeutic response and after disease relapse, and discovered a canonical cancer mutation PTEN R130* at both time points. The cellular and cfDNA revealed similar mutation profiles, suggesting cfDNA is representative of LMD cells. This study demonstrates the potential of using cellular or cfDNA in CSF to monitor treatment response for LMD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Leptomeningeal disease (LMD) is a distinct subtype of central nervous system (CNS) metastases, defined as the spread of tumor cells into the membranes surrounding the brain, spinal cord, and cranial nerves. LMD is diagnosed in approximately 5 % of patients with metastatic cancer, and is mostly commonly associated with breast cancer, lung cancer, and melanoma [1, 2]. Treatment of LMD may include radiation, intrathecal chemotherapy, or systemic chemotherapy. Regardless of treatment method, the prognosis remains poor, with survival ranging from weeks to months after diagnosis [1, 2]. Chemotherapy may not adequately penetrate the CNS, or LMD is treatment resistant. The lack of information regarding the genetic profile and molecular biology of LMD has significantly stymied research into an effective treatment of this disease [3, 4]. Conventional diagnostic approaches for LMD—CSF cytology and MRI imaging—have limited precision and accuracy, and frequently require multiple invasive procedures or studies to establish a definitive diagnosis. Even when positive, cytology and MRI do not provide quantitative analysis of treatment response, genomic information on targetable mutations, or insight into evolving tumor biology. CNS-penetrant therapies (e.g. erlotinib, dabrafenib) could be targeted to the LMD mutation profile (e.g. EGFR, BRAF), but until recently, a reliable method to test CSF for the genetic aberrations of brain tumors did not exist. Recent advances in the analyses of cell-free DNA (cfDNA), low level cellular DNA, and circulating tumor cells (CTCs), have made it possible to evaluate the various tumor mutation profiles [5]. This has important implications for guiding targeted therapeutics, accurately diagnosing and monitoring of LMD, and furthering our understanding of the genetic profile unique to LMD.
While systemic tumor cfDNA in human blood has great potential for cancer diagnosis, prognosis and treatment, this has not been demonstrated for plasma cfDNA in brain tumor patients [5, 6]. CSF from patients with LMD contains CTCs, and CSF contains cellular and cfDNA from metastatic and primary brain tumor cells, all of which can provide a less-invasive and routinely accessible method to obtain genomic information about brain tumors [5, 7, 8]. Although CTCs represent a useful source of LMD genetic information, they require a biased method of targeted capturing. In contrast, cellular and cfDNA can be easily isolated after centrifugation, and do not select cells based on a particular expression profile (e.g. EpCAM). Here we use tumor cellular and cfDNA from CSF to monitor the therapeutic response of a patient with LMD from a primary melanoma with a BRAF V600E mutation. We discuss the clinical significance of CSF examination using PCR-based techniques for accurate detection of characteristic mutations in LMD, the potential for monitoring response to therapy, and the potential to unveil key genes involved in this tumor’s malignant spread.
Methods and materials
Study design
A patient with elevated intracranial pressure from melanoma metastatic to the leptomeninges underwent serial therapeutic lumbar punctures (LPs). Patient samples were obtained through a Stanford University Hospital Institutional Review Board-approved informed consent process. Access of CSF was performed for clinical necessity and only excess CSF not required for pathologic diagnosis was utilized in this study; no procedures (e.g. LPs) were undergone for the exclusive purpose of research. CSF samples were always taken from the last tube of the procedure. Figure 1 depicts the overall study design. The blood and CSF samples were collected and centrifuged to separate cfDNA and cellular DNA. DNA was then extracted and prepared for droplet digital PCR (ddPCR) and exome sequencing, respectively, as described below.

Study design. Samples were collected from a patient undergoing sequential lumbar punctures for the diagnosis and treatment of melanoma leptomeningeal metastases (1). Samples were centrifuged to separate plasma from blood cells and CSF from tumor cells (2). After DNA extraction (3) cellular DNA from CSF and blood underwent exome sequencing to identify tumor mutations. cfDNA from plasma and CSF was utilized to determine the mutant fraction of the known BRAF mutation via ddPCR
Sample processing
CSF was spun down (1000 g, 10 min) within 2 h of collection. The supernatant containing cfDNA, and precipitant containing cellular DNA, were kept at −80 °C until ready for DNA extraction. The blood sample was separated into plasma and blood cells by centrifugation (1000 g, 10 min). The plasma went through another centrifugation (10,000 g 10 min) to remove residual cells. DNA from CSF was extracted with a QIAamp Circulating Nucleic Acid Kit (Qiagen), while DNA from blood cells was extracted with a QIAamp DNA Mini Kit (Qiagen). Cellular DNA, extracted from the precipitant of CSF or blood cells, was fragmented by sonication (S220 focused ultrasonicator, Covaris). The size distribution of DNA was measured by a fragment analyzer (Advanced Analytical).
Droplet digital PCR
The droplet digital PCR (ddPCR) assays were performed by a QX100TM Droplet DigitalTM PCR System (Biorad) following the manufacturer’s instructions. Each cfDNA sample was mixed with primers and fluorophore labeled probes (FAM for mutant, HEX for wild type). Cellular DNA was sheared into 300 base pair fragments by sonication before preparing the PCR mix. After PCR, the droplet reader detected fluorescent signals in each droplet. The concentration of the target mutant (C MUT ) and wild-type (C WT ) DNA in the PCR mix was calculated automatically by QuantaSoft software (Biorad) in copies/uL. The mutant allele fraction (MAF) was calculated for each sample with the following equation:
Concentrations of mutant and wild-type alleles were calculated with the following equations:
where C MUT_ori is the mutant allele concentration in the original CSF or plasma (copies/mL); C WT_ori is the wild-type allele concentration in the original CSF or plasma (copies/mL); V O is the original volume of CSF or plasma used for DNA extraction; V E is the elution volume generated from the DNA extraction; and V P is the volume of DNA solution used in final PCR mix. The volume of the final PCR mix was 20 μL.
Whole exome sequencing
Ten nanograms of cfDNA from CSF and 200 ng of matched normal DNA (from blood) were used to prepare indexed Illumina libraries. The DNA was fragmented to about 300 base pairs by sonication (S220 focused ultrasonicator, Covaris) before library construction. The Illumina libraries were constructed using KAPA Hyper Prep Kit (Kapa Biosystems). NimbleGen SeqCap EZ Human Exome Library (Roche) was used to capture DNA from exonic regions. Approximately 100 million reads per library were sequenced using 150-bp paired-end runs on NextSeq sequencing system (Illumina) and were analyzed as previously described [5]. Briefly, we first trimmed the raw FASTQ reads with Trimmomatic [9], and aligned the trimmed reads to human reference genome (hg19 assembly) with BWA software [10]. PCR duplicates were then removed using the Picard MarkDuplicates program, and reads were piped through GATK Indel Realignment and BaseRecalibration [11]. Somatic single-nucleotide variants (SNVs) were identified with MuTect [12] and annotated with ANNOVAR software [13].
Results
Clinical case
A 45-year-old patient with previously diagnosed, but unstaged, melanoma began experiencing headaches, nausea, and vomiting. An MRI of the brain showed abnormal contrast enhancement of right cranial nerves VII and VIII in the internal auditory canal (Fig. 2a; red arrow). LMD and increased intracranial pressure were confirmed by LP and cytology (Fig. 2b; blue arrow). The patient underwent whole brain radiation therapy with concurrent temozolomide (WBRT 37.5 Gy; TMZ 75 mg/m2; Fig. 3a, time point 1) for treatment of the LMD. The primary melanoma was BRAF positive and he was started on dabrafenib–trametinib (Fig. 3a, time point 2). The patient successfully responded to therapy and reported resolution of his symptoms approximately 2 months after beginning treatment (Fig. 3a, time point 6). Unfortunately, 3 months later the patient’s symptoms returned (Fig. 3a, time point 7), and despite ipilimumab therapy, he developed hydrocephalus for which he underwent ventriculoperitoneal shunt placement (Fig. 3a, time point 9).

a MRI brain, axial T1 with contrast, showing abnormal contrast enhancement of the right internal auditory canal (red arrow), consistent with cranial nerve involvement by leptomeningeal disease from metastatic melanoma. b CSF cytology (Papanicolaou stain) showing enlarged cell (blue arrow) with enlarged, eccentric nucleoli and intracytoplasmic pigment consistent with metastatic melanoma

The fraction of mutant BRAF cfDNA in CSF mirrored the clinical symptoms of LMD from metastatic melanoma. a The patient presented to clinic (1) with severe headaches, nausea, and vomiting. Cytology and MRI were consistent with LM metastases of his BRAF-mutated melanoma. The patient started whole brain radiation (RT) and temozolomide (2). He completed RT/temozolomide and began dabrafenib-trametinib, however, the patient’s symptoms persisted. As his symptoms began to improve (3–5), the fraction of mutant DNA in the CSF likewise decreased. When the patient was asymptomatic (6) the BRAF mutation was undetectable in CSF. As the patient was clinically well, additional lumbar punctures were not indicated (6–7). Ninety days later (7), the patient represented to clinic with severe headaches, nausea, and vomiting. He also developed hydrocephalus, requiring placement of a ventriculoperitoneal shunt (9). b Examples of ddPCR plot (time point 8) for CSF cfDNA showing a BRAF mutant fraction of 38 %. Mutants are clustered in the upper left corner with high FAM fluorescent intensities (blue), wild-type is clustered in lower right corner with high HEX fluorescent intensities (green). c Corresponding ddPCR plot (time point 8) for plasma cfDNA did not detect the BRAF mutation, indicating plasma cfDNA was not reflective of the tumor mutation profile within the CNS
Detection of tumor cfDNA in CSF
Melanoma routinely undergoes clinical mutation testing to determine the most appropriate targeted therapeutic. As a result, the primary melanoma tumor of our patient was known to have a BRAF point mutation 1799 T > A (V600E). The melanoma cells metastatic to the leptomeninges of the brain contained this same characteristic mutation, as detected with ddPCR in CSF on the day of cytologic diagnosis with the first LP (Fig. 3a, time point 1). Plasma cfDNA did not contain the mutant allele (Fig. 3c). The concentration and ratio of mutant DNA decreased as the patient’s clinical status improved following treatment for LMD, including whole brain radiation and dabrafenib-trametinib (Fig. 3a). As the patient’s symptoms of severe headaches, nausea, and vomiting resolved, the mutant fraction decreased progressively; the BRAF mutation was undetectable in CSF at the point of clinical recovery. As the CSF samples were collected only when a clinically indicated LP was performed, we do not have additional sample time points when the patient was not having symptoms (time point 6 to 7, Fig. 3a). After 3 months, when the patient returned with recurrence of his symptoms (headache, nausea, vomiting), the mutant tumor DNA was again detected in CSF at a high fraction (time point 7).
We compared the tumor cellular and cfDNA contributions in CSF (see supplementary Fig. S1). The cellular DNA from the 7th time point had a similar mutant allele fraction (54 %) as its cell-free compartment (60 %). In comparison to the mutant allele fraction, total cfDNA in CSF likewise followed the clinical response to therapy and relapse (see supplementary Fig. S2).
Exome sequencing
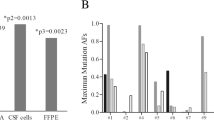
Exome sequencing was performed on DNA from CSF and blood cells from pretreatment (Fig. 3a, time point 1) and relapse (Fig. 3a, time point 8) to identify cancer mutations. The pretreatment sample was from cfDNA in CSF, with a limited amount of DNA available for sequencing (~5 ng). At relapse we were able to collect over 100 ng of CSF cellular DNA for sequencing. Not surprisingly, given the larger amount of DNA available for sequencing in the latter CSF sample, the relapse data had higher sequencing coverage and fewer artifacts as shown in Fig. 4. In both pretreatment and relapsed samples, 47 nonsynonymous SNV mutations appeared (highlighted in blue in Fig. 4; mutual). These mutations tended to have higher sequencing coverage and mutant allele frequency. The mutant allele reads were generally more than 15 in the pretreatment sample and 25 in the relapsed sample, increasing the likelihood that these were true mutations. Two canonical tumor mutations, BRAF V600E and PTEN R130*, were present in both samples (highlighted in red in Fig. 4; canonical). The other identified mutations (highlighted in grey in Fig. 4; nonmutual) have either low sequencing coverage or low tumor allele frequency, and are likely artifacts caused by sequencing error, PCR error, or misalignment. Several other mutations occurred in cancer census genes (http://cancer.sanger.ac.uk/cancergenome/projects/census/), but did not fall into canonic cancer mutation loci (pretreatment and relapse samples; Table 1). Of these mutations, none were shared between the pretreatment and relapse samples (included in grey mutations in Fig. 4). The mutant allele reads were all less than 10, making it difficult to draw any definitive conclusions. See tabulated data sheet in Supplementary Information 2.

Mutations identified by exome sequencing from pretreatment time point (left) and relapse time point (right). Mutual mutations are shown in blue, nonmutual mutations are shown in grey, and canonical cancer mutations are shown in red
Discussion
While current clinical cytology tests provide no genetic information of tumor cells and no quantitative assessment of disease severity (supplementary Table S1), cell-free DNA is a valuable tool in the diagnosis, prognosis, and monitoring of therapy response in various cancer types, such as breast, lung, ovarian, and prostate [14–17]. cfDNA for tumors in the CNS has rarely been studied due to the difficulty of detecting CNS tumor cfDNA in plasma [5, 18].We were unable to detect mutant cfDNA in the plasma (Fig. 3c) even though plasma contained a higher total concentration of cfDNA. We were, however, able to detect two distinct populations of mutant and wild-type cfDNA in CSF. Our inability to detect mutant cfDNA in plasma likely resulted from the active metastatic disease being confined to the CNS. Thus, CNS tumor DNA in CSF was more representative of the disease then could be determined in plasma, suggesting it as a useful tool for detailing the extent, progression, and genetic characteristics of metastatic CNS disease.
Our time-course analysis of CSF tumor cellular and cfDNA was highly correlated with therapeutic response and relapse. Besides the mutant allele fraction, the overall trend of total cfDNA suggested a higher cfDNA amount corresponded with more severe symptoms (see supplementary Fig. S2), however, the total cfDNA amount may have artifacts depending on sample handling, extraction process and storage, making it less reliable than the mutant fraction to monitor treatment response. As the samples were only collected during a clinically indicated LP, 3 months had elapsed between the patient’s symptom resolution and return. If we had CSF samples during that intervening period, prior to the time of clinical recurrence, could we have predicted the return of symptoms? Given the potential morbidity associated with an LP, without these preliminary data showing that the tumor mutant fraction correlates with clinical response and relapse, LPs for the express purpose of research would not have been appropriate. Although we have matched samples from the same patient who serves as his own control, this initial study follows only one patient; additional longitudinal studies with multiple samples from multiple patients is an important next step.
LMD from melanoma and other solid tumors is frequently treated with whole brain radiation, which merely delays disease progression and involves numerous deleterious side effects. Targeted therapies have shown promising responses for both parenchymal and leptomeningeal brain metastases [19, 20]. Vemurafenib has demonstrated improved survival in melanoma with BRAF V600E [21], and the use of BRAF kinase inhibitors for the treatment of brain metastases is currently under investigation in clinical trials [22–25]. In the future, digital PCR-based assessment of tumor DNA in CSF may allow for an objective assessment of a clinical response to therapy, and allow for a determination of a BRAF mutation in patients with isolated CNS disease, without the need for an invasive brain tumor tissue biopsy.
As is frequently the case in patients with metastatic melanoma, the initial primary tumor biopsy was done at an outside facility, tested for a BRAF mutation, and no tissue was available for comparison to our LMD samples. Ideally the cfDNA within CSF would be compared to a metastatic deposit in the CNS, however, this is not possible as such a procedure would be contraindicated and morbid. The biologic equivalent to open LM CNS tissue are the LM cells that shed themselves circulating within the CSF. We found that exome sequencing of the LM cell pellet (LM tissue diagnosis equivalent) was similar to exome sequencing of cfDNA from the same sample. This finding suggests that even when LM cells are not captured in a patient with LMD, tumor cfDNA in CSF would reliably reflect the genetic profile of LM tumor cells within the CNS.
While sample scarcity and low levels of DNA within CSF limited our capacity to perform exome sequencing on multiple time points, important information was gained from the samples obtained at presentation and recurrence. Similar mutation profiles in both data points suggest no substantial evolution of tumor cells after LMD diagnosis. Although the mutant allele fractions of BRAF V600E and PTEN R130* differed between the two time points, both increased about 25 % at the time of recurrence, conceptually supporting the existence of a single clonal tumor cell population. PTEN and BRAF genes are known to be common driver mutations in many cancers, and the loss of PTEN has cooperated with activation of BRAF in the progression [26] and metastasis [27] of melanoma. Moreover, PTEN loss plays a critical role in the resistance of BRAF inhibitor chemotherapy [28]. In this case, PTEN loss was present before targeted treatment, calling into question if the initial symptom alleviation may have been a response to whole brain radiation or the targeted therapeutics.
The characteristic pattern of LMD growth and poor prognosis suggests an underlying tumor genetic profile distinct from solid metastatic tumors. Next-generation sequencing techniques have allowed us to accurately sequence progressively smaller amounts of DNA, techniques necessary to process the scarce tumor cellular and cfDNA present in LMD. Exome sequencing of melanoma LMD revealed interesting mutations in non-cancer genes, which may be linked to the unique biology of LMD. For example, several genes involved in cell motility and adhesion were identified, and are also hypothesized to have an effect on cancer metastasis. ARHGAP30 is reported to play a role in cell migration, focal adhesion dissolution, and its ectopic expression results in membrane blebbing and dissolution of stress-fibers and focal adhesions [29]. Other genes encode important components related to cytoskeleton and cell plasticity, such as DRC7 [30] and LPPR1 [31]. Confirmation of the role of these genes in LMD requires further validation, including more robust analysis of LMD at the RNA level.
Conclusion
LMD tumor cellular and cfDNA from CSF can identify characteristic mutations that have clinical implications for selection of targeted therapeutics or to monitor treatment response. The mutant allele fraction in cfDNA quantified by ddPCR corresponds well with clinical response, and opens the possibility of broadening the scope of our project to additional patients with LMD from a variety of primary tumor sources. Our bulk exome sequencing results indicated that cfDNA in CSF can comprehensively reflect the LMD genomic profile. Future studies may investigate the transcriptome at the bulk and single-cell level to explore the mechanism of tumor metastasis and relapse.
References
Chamberlain MC (2010) Leptomeningeal metastasis. Curr Opin Oncol 22(6):627–635
Weller M (2003) Leptomeningeal metastasis. In: Brant T (ed) Neurological disorders: course and treatment, 2nd edn. Academic Press, Amsterdam, pp 897–909
Nagpal S, Riess J, Wakelee H (2012) Treatment of leptomeningeal spread of NSCLC: a continuing challenge. Curr Treat Options Oncol 13(4):491–504
Wasserstrom WR, Glass JP, Posner JB (1982) Diagnosis and treatment of leptomeningeal metastases from solid tumors: experience with 90 patients. Cancer 49(4):759–772
Pan W, Gu W, Nagpal S, Gephart MH, Quake SR (2015) Brain tumor mutations detected in cerebral spinal fluid. Clin Chem 61(3):514–522
Bettegowda C, Sausen M, Leary RJ et al (2014) Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 6(224):224ra224
Jang NE, Baek SK, Jeong Jh et al (2013) Early detection of BCR-ABL fusion gene of cerebrospinal fluid (CSF) by RT-PCR in relapsed acute lymphoblastic leukemia with philadelphia chromosome. Lab Med 43(2):e33–e37
Swinkels DW, de Kok JB, Hanselaar A, Lamers K, Boerman RH (2000) Early detection of leptomeningeal metastasis by PCR examination of tumor-derived K-ras DNA in cerebrospinal fluid. Clin Chem 46(1):132–133
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25(14):1754–1760
McKenna A, Hanna M, Banks E et al (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20(9):1297–1303
Cibulskis K, Lawrence MS, Carter SL et al (2013) Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31(3):213–219
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38(16):e164
De Mattos-Arruda L, Cortes J, Santarpia L et al (2013) Circulating tumour cells and cell-free DNA as tools for managing breast cancer. Nat Rev Clin Oncol 10(7):377–389
Kamat AA, Baldwin M, Urbauer D et al (2010) Plasma cell-free DNA in ovarian cancer: an independent prognostic biomarker. Cancer 116(8):1918–1925
Papadopoulou E, Davilas E, Sotiriou V et al (2006) Cell-free DNA and RNA in plasma as a new molecular marker for prostate and breast cancer. Ann N Y Acad Sci 1075:235–243
Pathak AK, Bhutani M, Kumar S, Mohan A, Guleria R (2006) Circulating cell-free DNA in plasma/serum of lung cancer patients as a potential screening and prognostic tool. Clin Chem 52(10):1833–1842
Wang Y, Springer S, Zhang M et al (2015) Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc Natl Acad Sci 112(31):9704–9709
Grommes C, Oxnard GR, Kris MG et al (2011) “Pulsatile” high-dose weekly erlotinib for CNS metastases from EGFR mutant non-small cell lung cancer. Neuro Oncol 13(12):1364–1369
Schulze B, Meissner M, Wolter M, Rodel C, Weiss C (2014) Unusual acute and delayed skin reactions during and after whole-brain radiotherapy in combination with the BRAF inhibitor vemurafenib. Two case reports. Strahlenther Onkol 190(2):229–232
Chapman PB, Hauschild A, Robert C et al (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364(26):2507–2516
Falchook GS, Long GV, Kurzrock R et al (2012) Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet 379(9829):1893–1901
Fennira F, Pages C, Schneider P et al (2014) Vemurafenib in the French temporary authorization for use metastatic melanoma cohort: a single-centre trial. Melanoma Res 24(1):75–82
Sosman JA, Kim KB, Schuchter L et al (2012) Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med 366(8):707–714
Wilgenhof S, Neyns B (2014) Complete cytologic remission of V600E BRAF-mutant melanoma-associated leptomeningeal carcinomatosis upon treatment with dabrafenib. J Clin Oncol 33:e109
Tsao H, Goel V, Wu H, Yang G, Haluska FG (2004) Genetic interaction between NRAS and BRAF mutations and PTEN/MMAC1 inactivation in melanoma. J Invest Dermatol 122(2):337–341
Dankort D, Curley DP, Cartlidge RA et al (2009) Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet 41(5):544–552
Paraiso KH, Xiang Y, Rebecca VW et al (2011) PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res 71(7):2750–2760
Naji L, Pacholsky D, Aspenstrom P (2011) ARHGAP30 is a Wrch-1-interacting protein involved in actin dynamics and cell adhesion. Biochem Biophys Res Commun 409(1):96–102
Yang Y, Cochran DA, Gargano MD et al (2011) Regulation of flagellar motility by the conserved flagellar protein CG34110/Ccdc135/FAP50. Mol Biol Cell 22(7):976–987
Strauss U, Brauer AU (2013) Current views on regulation and function of plasticity-related genes (PRGs/LPPRs) in the brain. Biochim Biophys Acta 1831(1):133–138
Acknowledgments
We recognize our patient’s generous gift of allowing us to collect and analyze genetic material from this fatal tumor, and thank Cindy H. Samos for manuscript editing.
Funding
This work was supported in part by HHMI (SQ, WP), NIH grant R21CA193046-01 (MHG), The Hearst Foundation (MHG), and a postdoctoral fellowship from Stanford Child Health Research Institute (YL).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors have no financial interests or any conflicts of interest to declare in the preparation of this work.
Additional information
Yingmei Li and Wenying Pan contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Li, Y., Pan, W., Connolly, I.D. et al. Tumor DNA in cerebral spinal fluid reflects clinical course in a patient with melanoma leptomeningeal brain metastases. J Neurooncol 128, 93–100 (2016). https://doi.org/10.1007/s11060-016-2081-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-016-2081-5