
Abstract
MicroRNAs (miRNAs) are small noncoding regulatory RNAs that reduce stability and/or translation of fully or partially sequence-complementary target mRNAs. Recent evidence indicates that miRNAs can function both as tumor suppressors and as oncogenes. It has been demonstrated that in glioblastoma multiforme miR-21 and 221 are upregulated whereas miR-128 and 181 are downregulated. Expression of miR-21, 221, 128a, 128b, 128c, 181a, 181b, 181c was studied using real-time quantitative reverse transcriptase polymerase chain reaction and northern blotting for human astrocytic tumors with different grade of malignancy. miR-21 and 221 were overexpressed in glioma samples, whereas miRNA 181b was downregulated compared with normal brain tissue. miRNA-21 was hyperexpressed in all tumor samples whereas higher levels of miRNA-221 were found in high-grade gliomas. This study is the first analysis of miRNAs in astrocytic tumor at different stages of malignancy. The different expression pattern observed in tumors at different stages of malignancy is probably dependent on the cell-specific repertoire of target genes of tumors sharing different molecular pathways activity and suggests miRNAs may have also a place in diagnosis and staging of brain tumors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
MicroRNAs (miRNAs) are a recently discovered class of small, evolutionary conserved, RNA molecules that negatively regulate gene expression at the post-transcriptional level. The discovery of these small noncoding transcripts has broadened our understanding of the mechanisms that regulate gene expression, adding an entirely novel level of regulatory control.
miRNAs consist of 18–25 nucleotides and are a class of endogenous ribo-regulators that modulate gene expression via the RNA interference (RNAi) pathway. RNAi is a post-transcriptional silencing mechanism, present in most eukaryotic organisms, in which exposure to double-stranded RNA induces the sequence-specific degradation of homologous messenger RNAs (mRNA). miRNAs act by base-pairing with their target mRNAs through perfect or nearly perfect complementarity particularly at the 3′ untranslated regions (UTRs) of the target mRNAs [1–5] leading to their translational repression and/or direct cleavage [6].
To understand the mechanism of miRNA-mediated silencing, the basic step of their biogenesis must be summarized. miRNAs originate from long primary miRNAs (pri-miRNAs) that are transcribed in the nucleus by the RNA polymerase II complex. miRNAs are then processed to 60–70-nucleotide precursor miRNA (pre-miRNA) intermediates by a complex of the RNase III enzyme DROSHA and a double-stranded RNA binding domain possessing the protein DGCR8 (DiGeorge syndrome critical region gene 8). These hairpin-shaped pre-miRNAs are transported to the cytoplasm by exportin-5 (Exp5, a member of the Ran transport receptor family). Once in the cytoplasm, they are cleaved by DICER to generate 20–22-nucleotide duplexes bearing two nucleotide single-stranded 3′ extensions. Finally, the miRNA–miRNA* duplex is unwound into mature miRNA and miRNA* by a helicase. Single-strand miRNA is incorporated into a ribonucleoprotein effector complex, known as the RNA-induced silencing complex (RISC), whereas miRNA* is degraded [7–9]. RISC identifies target messages based on complementarities between the “guide” miRNA and the mRNA, and results in either endonucleolytic cleavage of targeted mRNA or translational repression.
There are estimates, in part based on computational methods, that mammalian genomes encode up to 1,000 unique miRNAs [10], which are predicted to regulate expression of as much as 30% of genes [11]. Although more than 530 miRNAs have been identified in humans, much remains to be understood about their precise cellular function and role in the development of diseases. Because miRNAs are implicated in the control of many fundamental cellular and biological processes, for example developmental timing [12], stem cell division [13, 14], and apoptosis [15], changes in their expression may play a role in the development of diseases [16, 17] and cancer [6, 18]. Recent evidence indicate that miRNAs can function both as tumor suppressors and oncogenes [19, 20]. Indeed, altered miRNA expression has been reported in leukemia, lung cancer, colon cancer, and breast cancer [21–23], and in many other malignancies.
A role of miRNA has also been suggested in glioblastoma (GBM), the most common tumor of the brain and one of the most aggressive tumor in humans [24]. Ciafre et al. studied the global expression of 245 microRNAs in GBM using a microarray technique [25]. This approach enabled the identification of miRNAs whose expression is significantly altered in tumors compared with peripheral brain areas from the same patient, including miR-221, strongly up-regulated in glioblastoma, and a set of brain-enriched miRNAs, miR-128, miR-181a, miR-181b, and miR-181c, which were down-regulated in glioblastoma. Chan et al. [26] used oligonucleotide arrays specific for 180 human and mouse miRNAs and found marked elevation of miR-21 levels in human primary GBM tissues and cell lines.
Nevertheless, nothing is known about the expression of miRNAs in astrocytic tumors at earlier stages of malignancy. In this study we analyzed the expression of the abovementioned set of miRNAs in human astrocytic tumors at different stages of malignancy.
Material and methods
Patient population
This study included tumor samples histologically verified as grades II–IV astrocytomas obtained from adult patients who had undergone craniotomy for microsurgical tumor removal. All tumors were located in the supratentorial compartment. Only patients who had undergone extensive gross-total resection of the neoplasm (95% of the tumor volume) were eligible to participate in the study. We excluded oligodendroglial tumors, recurrent tumors, and patients who had undergone neoadjuvant therapy (radiotherapy and/or chemotherapy) before surgery. Furthermore, four samples of normal brain tissue were used as controls.
The study included 16 men and 12 women whose mean age was 55.4 ± 16.5 years at surgery (range 27–76 years). The Karnofsky performance status (KPS) scores in these patients was 91.1 ± 12.6 (range 60–100). According to the revised World Health Organization (WHO) classification [27], tumors were diagnosed as low-grade astrocytoma (LGA; eight patients), anaplastic astrocytoma (AA; ten patients), and glioblastoma multiforme (GBM; ten patients). Patients with LGAs had a mean age of 37.1 ± 6 years, a KPS score of 100, and a survival period longer than 208 weeks. Patients with AAs had a mean age of 62.1 ± 15.4 years, a mean KPS score of 86 ± 16.5, and a mean survival period of 109.3 ± 17.9 weeks (median 112 weeks). Patients with GBMs had a mean age of 51.4 ± 12.2 years, a mean KPS score of 89 ± 9.9, and a mean survival period of 51.4 ± 9.9 weeks (median 54 weeks). The total duration of follow up was 208 weeks postsurgery.
Tissue samples
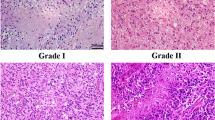
All tumor tissue samples were obtained from the neoplastic tissue within 15 min of surgical removal. Neoplastic tissue samples were taken from viable areas of tumor, while trying to avoid areas of gross necrosis. Three anatomically separate areas of the tumor were sampled from each resected specimen. Non-neoplastic brain tissue samples were derived from the temporal lobes of patients surgically treated for temporal lobe epilepsy and included histologically verified normal cortex and white matter. Tissue samples for analysis were placed in cryovials and immediately flash frozen in liquid nitrogen in the operating room and stored at −80°C. Tissue adjacent to the frozen samples and additional tissue obtained from the resection specimens were formalin-fixed, paraffin-embedded, cut, and stained with H & E and used for histological typing and grading on the basis of the WHO criteria.
RNA extraction
Enriched microRNAs were extracted using the mirVana miRNA Isolation kit (Ambion, Italy), and total RNA was also extracted using a total nucleic acid Isolation kit (Ambion) and following the manufacturer’s procedure. The concentrations of samples were measured spectrophotometrically and the quality was checked on agarose gel.
Reverse transcriptase reactions
Reverse transcriptase reaction contained RNA samples including purified miRNA, 50 nM stem-loop RT primer of each miRNA (RNU6, 21, 128a, 128b, 128c, 181a, 181b, 181c, and 221; purchased from Applera, Italy), 0.25 mM each of dNTPs, 3.33 U/μl MultiScribe reverse transcriptase (P/N: 4319983; Applied Biosystems, Milan, Italy) and 0.25 U/μl RNase inhibitor (P/N: N8080119; Applied Biosystems, Milan, Italy). The 7.5 μl reactions were incubated in an thermocycler for 30 min at 16°C, 30 min at 42°C, 5 min at 85°C, and then held at 4°C. All reverse transcriptase reactions, including no-template controls and RT minus controls, were run in duplicate.
Real-time PCR
Real-time PCR was performed using a standard TaqMan PCR kit procedure on an Applied Biosystems 7300 real-time PCR system (Applied Biosystems). The 10 μl PCR included 0.67 μl RT product, 1× TaqMan Universal PCR Master Mix (P/N: 4324018, Applied Biosystems), and 0.2 μM TaqMan probe. The reactions were incubated in a 96-well plate at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. All reactions were run in triplicate. The threshold cycle (C T) is defined as the fractional cycle number at which the fluorescence passes the fixed threshold. TaqMan C T values were converted into absolute copy numbers using a standard curve from miRNA U6.
Northern blot analysis
RNA samples (10 μg each) were electrophoresed on 15% acrylamide and 7 mol/l urea Criterion precast gels (Bio-Rad) and transferred on to Hybond N+ membrane (Amersham Biosciences). Membranes were hybridized with oligonucleotide probes corresponding to the complementary sequences of the below indicated mature miRNAs. The oligo probes were designed on the basis of individual miRNA sequence information deposited in miRBase (http://microrna.sanger.ac.uk):
-
miR-21, 5′-ATCGAATAGTCTGACTACAACT-3′,
-
miR-128-a, 5′-AGTGTCACTTGGCCAGAGAAAA-3′,
-
miR-128-b, 5′-AGTGTCACTTGGCCAGAGAAAG-3′,
-
miR-128-c, 5′-TTGTAAGTTGGACAGCCACTCA-3′,
-
miR-181-a, 5′-TTGTAAGTTGCGACAGCCACTCA-3′,
-
mir-181-b, 5′-TTGTAAGTAACGACAGCCACCC-3′,
-
mir-181-c, 5′-AACAUUCAACCUGUCGGUGAGU-3′ and
-
miR-221, 5′-GAAACCCAGCAGACAATGTAGCT-3′.
Probes were 5′-end labeled using the polynucleotide kinase in the presence of [γ-32P]ATP. Hybridization was performed at 37°C in 7% SDS/0.2 mol/l Na2PO4 (pH 7.0) for 16 h. Membranes were washed at 42°C, twice with 2× standard saline phosphate (0.18 mol/l NaCl/10 mmol/l phosphate, pH 7.4), 1 mmol/l EDTA (saline-sodium phosphate-EDTA; SSPE), and 0.1% SDS, and twice with 0.5× SSPE/0.1% SDS. Northern blots were rehybridized after stripping the oligonucleotides used as probes in boiling 0.1% SDS for 10 min. As a control for normalization of RNA expression levels, we hybridized blots with an oligonucleotide probe complementary to the U6 RNA (5′-GCAGGGGCCATGCTAATCTTCTCTGTATCG-3′). After blotting, results were visualized on Kodak X-ray film (Kodak, Milan, Italy) by autoradiography.
Statistical data analysis
Statistical analysis was accomplished using the unpaired Student t-test with Welch correction to compare the expression levels of miR-21, 221, 128a, 128b, 128c, 181a, 181b, and 181c as quantified on real time RT-PCR in LGAs, AAs, and GBMs. Data analysis was performed with INSTAT, version 3.0, and PRISM, version 4.0 (Graphpad, San Diego, CA, USA).
Results
To identify deregulated miRNAs we used quantitative real time-PCR (Taqman) to measure expression of eight mature miRNA sequences in human non-neoplastic brain tissues (NBT), LGAs (WHO grade II); AAs (grade III), and GBMs (grade IV). The comparative Ct (ΔΔCt) method was used to determine the change in expression of each miRNA in tumor samples relative to that in normal brain tissue. Briefly, the ΔCt of each miRNA was determined relative to U6 endogenous control RNA, which was robustly and invariantly expressed across all samples, and the average ΔCt of the four NBT samples was used as the calibrator for the tumor samples.
miR-21 was up-regulated seven to elevenfold in tumor samples. In detail miRNA-21 changes over NBT were: 8.76 ± 1.25-fold in LGAs, 9.39 ± 1.46-fold in AAs, and 9.18 ± 2.54-fold in GBMs (Fig. 1a).
miR-221 was consistently upregulated in our sample set. miRNA-221 was up-regulated 5 to 17-fold in tumor samples. In detail, miRNA-21 changes over NBT were: 7.69 ± 1.43-fold in LGAs, 9.1 ± 2.4-fold in AAs, and 12 ± 2.5-fold in GBMs (Fig. 1b).
Expression of miR-181b decreased by a factor of 6 to 17. In detail, miRNA-181b changes over NBT were: a factor of 0.1 ± 0.03 in LGAs, a factor of 0.13 ± 0.03 in AAs, and a factor of 0.09 ± 0.03 in GBMs (Fig. 1c).
Analysis of miRNA-128a, 128b, 128c, 181a, 181c showed expression levels were within threefold those of NBT and were considered not significantly changed.
We next performed statistical analyses of our miRNA expression data to identify differences in expression among tumor sample with different grades of malignancy. There were no differences in expression levels of miRNA-21 and miRNA-181b among LGAs, AAs, and GBMs. miRNA-221 was significantly more up-regulated in GBMs compared with LGAs (P < 0.01) and AAs (P < 0.01) (Fig. 1d).

Real-time quantitative RT-PCR expression was performed on cDNA from patient samples for miR-21, miR-221, and miR-181b. The expression levels of each gene were calculated as multiples of their expression in normal brain tissue and depicted for each individual sample per disease type. a miR-21 was up-regulated 7 to 11-fold in tumor samples. b miRNA-221 was up-regulated 5 to 17-fold. c miR-181b expression was down-regulated by a factor of 6 to 17 in tumors compared with NBT. d Global representation of deregulated miRNA in the three different histotypes is also displayed. *P < 0.05; **P < 0.01. LGA, low-grade glioma; AA, anaplastic astrocytoma; GBM, glioblastoma multiforme
miRNAs were further assayed by Northern blot for validation purposes (Fig. 2). miRNAs expression was normalized with the corresponding U6 RNA in each sample. Northern blot analysis confirmed the results obtained by quantitative RT-PCR displaying similar expression pattern. In particular, northern blot analysis revealed that miR-21 was up-regulated in all glioma samples. miR-221 was significantly up-regulated in GBMs compared with LGAs.

Representative northern blot autoradiographs for internal control RNA U6 (upper), miRNA-21 (middle), and miRNA-221 (lower) expression studied in normal brain tissue and human gliomas. NBT, normal brain tissue; LGA, low-grade glioma; AA, anaplastic astrocytoma; GBM, glioblastoma multiforme
Discussion
Our results showed that miRNA 21 and 221 are up-regulated in human astrocytic tumors, whereas miRNA 181b is down-regulated. miRNA-21 was homogeneously overexpressed in low and high-grade tumors whereas miRNA-221 overexpression was more evident in high-grade tumors.
The discovery of miRNAs dates back to 1993 when Lee et al. described a small RNA, lineage-deficient-4 (lin-4), with antisense complementarity to lin-14 involved in the regulation of developmental timing in Caenorhabditis elegans [28]. At that time most investigators considered this small RNA as an oddity in worm genetics, but hundreds of these miRNAs were soon discovered in C. elegans and other animals by different laboratories [29–32]. There is currently evidence of expression of 340, 303, and 205 distinct mature miRNAs in human, mouse, and rat encoded by 395, 363, and 231 different miRNA genes, respectively [33].
MicroRNAs may play a role in the tumorigenesis and progression of cancer. Calin et al. first established a connection between microRNAs and cancer by showing that miR-15 and miR-16 are located on chromosome 13q14, a region deleted in more than half of B-cell chronic lymphocytic leukemia (CLL) [34]. Cimmino et al. then demonstrated that expression of miR-15a and miR-16-1 was inversely correlated with Bcl-2 expression in CLL and that both miRNAs negatively regulated Bcl-2 at a posttranscriptional level. Furthermore, Bcl-2 repression by these miRNAs induced apoptosis in a leukemic cell line model [15].
Our results agree with previous observations that miRNA-21 is overexpressed in some cancer histotypes, including gliomas. MicroRNA-21 is overexpressed in breast cancer [35, 36], and pancreatic cancer [37]. The first study on the possible involvement of miRNA in glioma revealed hyperexpression of miRNA-21 [26]. It showed markedly elevated miR-21 levels in human glioblastoma tumor tissues, in early-passage glioblastoma cultures, and in six established glioblastoma cell lines (A172, U87, U373, LN229, LN428, and LN308) compared with nonneoplastic fetal and adult brain tissues and compared with cultured nonneoplastic glial cells. Furthermore, the authors demonstrated that knockdown of miR-21 in cultured glioblastoma cells triggered the activation of caspases leading to increased apoptosis [26]. We observed that miRNA-21 is significantly upregulated in LGAs. These tumors are characterized by a low rate of proliferation and prolonged life-span, which suggests that defective apoptosis pathways may play a prominent role in the pathogenesis of such tumors.
Even though an antiapoptotic role is likely for miR-21 in gliomas, other mechanisms are possible, because in Hela cells its down-regulation determines, instead, an increase in cell proliferation without affecting apoptosis [38]. Meng et al. demonstrated that knock-down of miR-21 may render cholangiocarcinoma cells sensitive to gemcitabine whereas transfection of non-malignant cholangiocytes with the precursor of miR-21 renders them more resistant to this drug [39]. This may be the result of downregulation of PTEN, the phosphatase that antagonizes the growth-promoting activity of the PI-3 kinase-Akt signaling pathway. It is worthy of note that PTEN was originally cloned as a tumor suppressor for brain tumors with point mutations occurring in 25% of cases [40].
Further work by Frankel et al. on breast cancer cells [41] and by Asangani et al. [42] on colon cancer cells found that miR-21 overexpression leads to PDCD4 (programmed cell death 4) reduction, by direct interaction with its 3′ UTR, and that anti-miR-21 treatment is followed by an increase in endogenous PDCD4 protein levels. PDCD4 is a tumor suppressor known to be up-regulated during apoptosis [43] and reduced in different tumors [44], and evidence demonstrates that miR-21 overexpression effects are at least in part due to PDCD4 downregulation. Although this target has not been proven in brain tumors, it is possible that it may be relevant in these tumor types also, as a recent study demonstrated that most of the glioma samples analyzed lacked PDCD4 protein expression whereas adjacent normal glial tissues expressed high levels of PDCD4 [44]. An additional molecular mechanism, proving the oncogenic properties of miR-21 in an in-vivo xenograft breast cancer model, consists in targeting of the tumor suppressor protein tropomyosin 1 (TPM1) at the translational level. Tropomyosin 1 is able per se to reduce tumor cell proliferation and anchorage independence, therefore explaining the effects on tumor growth exerted by miR-21 [45].
A recent publication explored the expression of miRNAs in GBM, using microarray analysis to determine the global expression of miRNAs in tumors relative to normal brain [25]. Ciafre et al. confirmed the upregulation of miR-21, but also identified an additional set of upregulated miRNAs. Notably, amongst these was miR-221, which was up-regulated in a subset of glioblastoma samples and in all glioblastoma cell lines examined. Our results confirm the hyperexpression of miR-221 in glioma.
The role of miR-221 in tumor development has been also analyzed. Gillies and Lorimer defined a specific function of miRNA 221 and 222 in glioblastoma, showing that they repress expression of the cell cycle regulatory protein p27Kip1 [46]. The p27Kip1 gene is a member of the Cip/Kip family of cyclin-dependent kinase (CDK) inhibitors that function to negatively control cell cycle progression. The protein binds to CDK2 and cyclin E complexes to prevent cell cycle progression from G1 to S phase. p27Kip1 also acts as a tumor suppressor and its expression is often disrupted in human cancers. Analysis of the 3′ UTR of p27Kip1 suggests that repression of this protein is a consequence of direct binding of miRNA 221 and 222 to sites in the 3′ UTR.
This mechanism was suggested contemporaneously and independently by le Sage et al. who demonstrated, using miRNA inhibitors, that some glioblastoma cell lines require high activity of miR-221 and 222 to maintain low p27Kip1 levels and continuous proliferation, and that high levels of miR-221 and 222 seem to correlate with low levels of p27Kip1 protein in GBM [47].
Gonzalez et al. have previously shown that inhibition of cdk4 activity enhances translation of p27Kip1, providing a link between these two cell-cycle regulators [48]. This effect was shown to be mediated by the 3′ UTR of p27Kip1. The glioblastoma cells used in that study were mutated at the INK4A/ARF locus and did not express the cdk4 inhibitor p16Ink4a [49]. This suggested the possibility that loss of cdk4 inhibition through INK4a/ARF mutations causes an increase in miR-221 levels, and a consequent decrease in p27Kip1 levels.
According to those observations, the up-regulation of miRNA-221 in high-grade gliomas, recorded in our study, may suggest a specific role in the defective cell cycle control and high proliferation rate of those tumors compared with less malignant tumors. p27Kip1 is a well-known tumor suppressor down-regulated in many human tumors, and its expression levels in primary cancers are highly correlated with reduced patient survival [50]. Similarly, miR-221 has also been associated with poor prognostic miRNA signatures in thyroid papillary carcinomas [51, 52], pancreatic adenocarcinoma, and GBM [25, 37], indeed reinforcing interconnection between miR-221 and p27Kip1 in the pathogenesis of GBM.
Ciafre et al. in their microarray analysis of the global expression of miRNAs in glioblastoma found that miRNA-181a and miRNA-181b were downregulated in glioma specimens compared with non-tumoral adjacent tissue and in glioma cell lines compared with normal brain tissue [25]. There is limited but substantial evidence regarding the role of miRNA-181 in cancer. Cheng et al. showed that inhibition of miRNA-181a increases cell growth in the lung carcinoma cell line, A549 [53]. Pekarsky et al. found miRNA-181b was down-regulated in aggressive B-cell chronic lymphocytic leukemia with 11q deletion compared with the indolent form of chronic lymphocytic leukemia [54]. All this evidence seem to suggest that downregulation of miRNA-181 may play a role in cancer development. Very recently, Shi et al. reported the downregulation of miR-181a and 181b in both human gliomas and glioma cell lines, confirming the data of Ciafre et al. [55]. Those authors showed that transfection of those miRNAs triggered growth inhibition, apoptosis, and inhibited invasion. Furthermore, miR-181a expression turned out to be inversely correlated with tumor grading whereas miR-181b was uniformly downregulated in gliomas with different grades of malignancy. Our results partially confirm those observations and show significant and uniform downregulation of the miRNA-181b in human gliomas, with miRNA-181a showing a tendency toward downregulation which did not reach statistical significance. This difference was possibly because of the threshold selected to define the overexpression of miRNA, which was higher our study. Nonetheless, our results are based on a relatively small number of tumor samples, this limits their applicability and requires further studies.
Conclusions
In this study, we carried out an analysis of a set of miRNAs in astrocytic tumors at different stages of malignancy. Our results suggest that miRNA-21 is upregulated even in low-grade tumors whereas miRNA-221 upregulation may be a molecular mechanism characteristic of a higher grade of malignancy. The different expression pattern observed in tumors at different stages of malignancy is likely to be dependent on the cell-specific repertoire of target genes of tumors sharing different molecular pathways activity and suggests miRNAs may also have a place in diagnosis and staging of brain tumors. The demand for reliable markers suitable for prediction of prognosis is high, especially for cancer types such as gliomas. Further studies to fully elucidate the expression patterns of miRNAs in astrocytic tumors with different grades of malignancy are warranted.
References
de Moor CH, Meijer H, Lissenden S (2005) Mechanisms of translational control by the 3′ UTR in development and differentiation. Semin Cell Dev Biol 16:49–58. doi:10.1016/j.semcdb.2004.11.007
Lai EC (2002) Micro RNAs are complementary to 3′ UTR sequence motifs that mediate negative post-transcriptional regulation. Nat Genet 30:363–364. doi:10.1038/ng865
Robins H, Press WH (2005) Human microRNAs target a functionally distinct population of genes with AT-rich 3′ UTRs. Proc Natl Acad Sci USA 102:15557–15562. doi:10.1073/pnas.0507443102
Stark A, Brennecke J, Bushati N, Russell RB, Cohen SM (2005) Animal microRNAs confer robustness to gene expression and have a significant impact on 3′UTR evolution. Cell 123:1133–1146. doi:10.1016/j.cell.2005.11.023
Sun M, Hurst LD, Carmichael GG, Chen J (2005) Evidence for a preferential targeting of 3′-UTRs by cis-encoded natural antisense transcripts. Nucleic Acids Res 33:5533–5543. doi:10.1093/nar/gki852
Meltzer PS (2005) Cancer genomics: small RNAs with big impacts. Nature 435:745–746. doi:10.1038/435745a
Ro S, Park C, Young D, Sanders KM, Yan W (2007) Tissue-dependent paired expression of miRNAs. Nucleic Acids Res 35:5944–5953. doi:10.1093/nar/gkm641
Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ (2004) Processing of primary microRNAs by the Microprocessor complex. Nature 432:231–235. doi:10.1038/nature03049
Hammond SM, Bernstein E, Beach D, Hannon GJ (2000) An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 404:293–296. doi:10.1038/35005107
Berezikov E, Guryev V, van de Belt J, Wienholds E, Plasterk RH, Cuppen E (2005) Phylogenetic shadowing and computational identification of human microRNA genes. Cell 120:21–24. doi:10.1016/j.cell.2004.12.031
Lewis BP, Burge CB, Bartel DP (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120:15–20. doi:10.1016/j.cell.2004.12.035
Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G (2000) The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403:901–906. doi:10.1038/35002607
Cheng LC, Tavazoie M, Doetsch F (2005) Stem cells: from epigenetics to microRNAs. Neuron 46:363–367. doi:10.1016/j.neuron.2005.04.027
Hatfield SD, Shcherbata HR, Fischer KA, Nakahara K, Carthew RW, Ruohola-Baker H (2005) Stem cell division is regulated by the microRNA pathway. Nature 435:974–978. doi:10.1038/nature03816
Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, Rassenti L, Alder H, Volinia S, Liu CG, Kipps TJ, Negrini M, Croce CM (2005) miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA 102:13944–13949. doi:10.1073/pnas.0506654102
Abelson JF, Kwan KY, O’Roak BJ, Baek DY, Stillman AA, Morgan TM, Mathews CA, Pauls DL, Rasin MR, Gunel M, Davis NR, Ercan-Sencicek AG, Guez DH, Spertus JA, Leckman JF, Dure LS, Kurlan R, Singer HS, Gilbert DL, Farhi A, Louvi A, Lifton RP, Sestan N, State MW (2005) Sequence variants in SLITRK1 are associated with Tourette’s syndrome. Science 310:317–320. doi:10.1126/science.1116502
Alvarez-Garcia I, Miska EA (2005) MicroRNA functions in animal development and human disease. Development 132:4653–4662. doi:10.1242/dev.02073
Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, Prueitt RL, Yanaihara N, Lanza G, Scarpa A, Vecchione A, Negrini M, Harris CC, Croce CM (2006) A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA 103:2257–2261. doi:10.1073/pnas.0510565103
Dalmay T (2008) MicroRNAs and cancer. J Intern Med 263:366–375. doi:10.1111/j.1365-2796.2008.01926.x
Cho WC (2007) OncomiRs: the discovery and progress of microRNAs in cancers. Mol Cancer 6:60. doi:10.1186/1476-4598-6-60
Calin GA, Liu CG, Sevignani C, Ferracin M, Felli N, Dumitru CD, Shimizu M, Cimmino A, Zupo S, Dono M, Dell’Aquila ML, Alder H, Rassenti L, Kipps TJ, Bullrich F, Negrini M, Croce CM (2004) MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc Natl Acad Sci USA 101:11755–11760. doi:10.1073/pnas.0404432101
Michael MZ, O’ Connor SM, van Holst Pellekaan NG, Young GP, James RJ (2003) Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol Cancer Res 1:882–891
Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, Harano T, Yatabe Y, Nagino M, Nimura Y, Mitsudomi T, Takahashi T (2004) Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res 64:3753–3756. doi:10.1158/0008-5472.CAN-04-0637
Nicoloso MS, Calin GA (2008) MicroRNA involvement in brain tumors: from bench to bedside. Brain Pathol 18:122–129. doi:10.1111/j.1750-3639.2007.00119.x
Ciafre SA, Galardi S, Mangiola A, Ferracin M, Liu CG, Sabatino G, Negrini M, Maira G, Croce CM, Farace MG (2005) Extensive modulation of a set of microRNAs in primary glioblastoma. Biochem Biophys Res Commun 334:1351–1358. doi:10.1016/j.bbrc.2005.07.030
Chan JA, Krichevsky AM, Kosik KS (2005) MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res 65:6029–6033. doi:10.1158/0008-5472.CAN-05-0137
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114:97–109. doi:10.1007/s00401-007-0243-4
Lee RC, Feinbaum RL, Ambros V (1993) The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75:843–854. doi:10.1016/0092-8674(93)90529-Y
Pasquinelli AE, Reinhart BJ, Slack F, Martindale MQ, Kuroda MI, Maller B, Hayward DC, Ball EE, Degnan B, Muller P, Spring J, Srinivasan A, Fishman M, Finnerty J, Corbo J, Levine M, Leahy P, Davidson E, Ruvkun G (2000) Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature 408:86–89. doi:10.1038/35040556
Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T (2001) Identification of novel genes coding for small expressed RNAs. Science 294:853–858. doi:10.1126/science.1064921
Lau NC, Lim LP, Weinstein EG, Bartel DP (2001) An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 294:858–862. doi:10.1126/science.1065062
Lee RC, Ambros V (2001) An extensive class of small RNAs in Caenorhabditis elegans. Science 294:862–864. doi:10.1126/science.1065329
Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, Lin C, Socci ND, Hermida L, Fulci V, Chiaretti S, Foa R, Schliwka J, Fuchs U, Novosel A, Muller RU, Schermer B, Bissels U, Inman J, Phan Q, Chien M, Weir DB, Choksi R, De Vita G, Frezzetti D, Trompeter HI, Hornung V, Teng G, Hartmann G, Palkovits M, Di Lauro R, Wernet P, Macino G, Rogler CE, Nagle JW, Ju J, Papavasiliou FN, Benzing T, Lichter P, Tam W, Brownstein MJ, Bosio A, Borkhardt A, Russo JJ, Sander C, Zavolan M, Tuschl T (2007) A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 129:1401–1414. doi:10.1016/j.cell.2007.04.040
Calin GA, Croce CM (2006) MicroRNA signatures in human cancers. Nat Rev Cancer 6:857–866. doi:10.1038/nrc1997
Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, Menard S, Palazzo JP, Rosenberg A, Musiani P, Volinia S, Nenci I, Calin GA, Querzoli P, Negrini M, Croce CM (2005) MicroRNA gene expression deregulation in human breast cancer. Cancer Res 65:7065–7070. doi:10.1158/0008-5472.CAN-05-1783
Si ML, Zhu S, Wu H, Lu Z, Wu F, Mo YY (2007) miR-21-mediated tumor growth. Oncogene 26:2799–2803. doi:10.1038/sj.onc.1210083
Lee EJ, Gusev Y, Jiang J, Nuovo GJ, Lerner MR, Frankel WL, Morgan DL, Postier RG, Brackett DJ, Schmittgen TD (2007) Expression profiling identifies microRNA signature in pancreatic cancer. Int J Cancer 120:1046–1054. doi:10.1002/ijc.22394
Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, Lao KQ, Livak KJ, Guegler KJ (2005) Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res 33:e179. doi:10.1093/nar/gni178
Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T (2007) MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 133:647–658. doi:10.1053/j.gastro.2007.05.022
Ohgaki H, Kleihues P (2007) Genetic pathways to primary and secondary glioblastoma. Am J Pathol 170:1445–1453. doi:10.2353/ajpath.2007.070011
Frankel LB, Christoffersen NR, Jacobsen A, Lindow M, Krogh A, Lund AH (2008) Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J Biol Chem 283:1026–1033. doi:10.1074/jbc.M707224200
Asangani IA, Rasheed SA, Nikolova DA, Leupold JH, Colburn NH, Post S, Allgayer H (2008) MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 27:2128–2136. doi:10.1038/sj.onc.1210856
Zhang H, Ozaki I, Mizuta T, Hamajima H, Yasutake T, Eguchi Y, Ideguchi H, Yamamoto K, Matsuhashi S (2006) Involvement of programmed cell death 4 in transforming growth factor-beta1-induced apoptosis in human hepatocellular carcinoma. Oncogene 25:6101–6112. doi:10.1038/sj.onc.1209634
Gao F, Zhang P, Zhou C, Li J, Wang Q, Zhu F, Ma C, Sun W, Zhang L (2007) Frequent loss of PDCD4 expression in human glioma: possible role in the tumorigenesis of glioma. Oncol Rep 17:123–128
Zhu S, Si ML, Wu H, Mo YY (2007) MicroRNA-21 targets the tumor suppressor gene tropomyosin 1 (TPM1). J Biol Chem 282:14328–14336. doi:10.1074/jbc.M611393200
Gillies JK, Lorimer IA (2007) Regulation of p27Kip1 by miRNA 221/222 in glioblastoma. Cell Cycle 6:2005–2009
le Sage C, Nagel R, Egan DA, Schrier M, Mesman E, Mangiola A, Anile C, Maira G, Mercatelli N, Ciafre SA, Farace MG, Agami R (2007) Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J 26:3699–3708. doi:10.1038/sj.emboj.7601790
Gonzalez T, Seoane M, Caamano P, Vinuela J, Dominguez F, Zalvide J (2003) Inhibition of Cdk4 activity enhances translation of p27kip1 in quiescent Rb-negative cells. J Biol Chem 278:12688–12695. doi:10.1074/jbc.M207530200
Ishii N, Maier D, Merlo A, Tada M, Sawamura Y, Diserens AC, Van Meir EG (1999) Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol 9:469–479
Slingerland J, Pagano M (2000) Regulation of the cdk inhibitor p27 and its deregulation in cancer. J Cell Physiol 183:10–17. doi:10.1002/(SICI)1097-4652(200004)183:1<10::AID-JCP2>3.0.CO;2-I
He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, Hammond SM (2005) A microRNA polycistron as a potential human oncogene. Nature 435:828–833. doi:10.1038/nature03552
Pallante P, Visone R, Ferracin M, Ferraro A, Berlingieri MT, Troncone G, Chiappetta G, Liu CG, Santoro M, Negrini M, Croce CM, Fusco A (2006) MicroRNA deregulation in human thyroid papillary carcinomas. Endocr Relat Cancer 13:497–508. doi:10.1677/erc.1.01209
Cheng AM, Byrom MW, Shelton J, Ford LP (2005) Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res 33:1290–1297. doi:10.1093/nar/gki200
Pekarsky Y, Santanam U, Cimmino A, Palamarchuk A, Efanov A, Maximov V, Volinia S, Alder H, Liu CG, Rassenti L, Calin GA, Hagan JP, Kipps T, Croce CM (2006) Tcl1 expression in chronic lymphocytic leukemia is regulated by miR-29 and miR-181. Cancer Res 66:11590–11593. doi:10.1158/0008-5472.CAN-06-3613
Shi L, Cheng Z, Zhang J, Li R, Zhao P, Fu Z, You Y (2008) hsa-mir-181a and hsa-mir-181b function as tumor suppressors in human glioma cells. Brain Res 1236:185–193. doi:10.1016/j.brainres.2008.07.085
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Conti, A., Aguennouz, M., La Torre, D. et al. miR-21 and 221 upregulation and miR-181b downregulation in human grade II–IV astrocytic tumors. J Neurooncol 93, 325–332 (2009). https://doi.org/10.1007/s11060-009-9797-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-009-9797-4