
Abstract
Gliosarcoma (GS) is a glioblastoma with a sarcomatous component that is presumed to be a metaplastic differentiation of glioma cells. We studied the clinical relevance of this histological glioblastoma subentity within the pediatric population. We obtained patient data from the German HIT-GBM database, which contains clinical data for more than 600 pediatric patients with centrally reviewed high-grade gliomas. By applying defined inclusion criteria (diagnosis of GS proven by central neuropathological review; patient age 0 to 21 years), four patients were identified. In addition, after a review of the English medical scientific literature, 19 additional cases were found. The relative frequency of GS in the German HIT-GBM database was only 1.9%. In the whole series of 23 pediatric GS patients, including previously reported cases, the male-to-female-ratio was 1.2:1. GS was found in all pediatric age groups with a median age of 11 years, but there was an unexpectedly high accumulation in infants (6 of 23 <3 years of age, 26%). GS showed a strong predilection of the cerebral hemispheres (22 out of 23 cases). Increased intracranial pressure was the leading symptom of a short clinical history with a median duration of 0.7 month. Interestingly, six patients (26%) were reported with a history of cranial radiotherapy prior to GS diagnosis. In 60% of the GS patients in our series, gross total resection was achieved. Median overall (OS) and event-free survivals (EFS) of the total cohort were 12.1 and 9.8 months, respectively. In conclusion, GS is a very rare tumor entity in children. Literature review suggests a relatively higher incidence in infants and in patients with a previous history of radiotherapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gliosarcoma (GS; ICD-O code 9442/3) is a rare brain tumor characterized by histomorphologic heterogeneity, with alternating areas of glial and mesenchymal differentiation (Fig. 1) [1]. According to the most recent World Health Organization (WHO) classification of central nervous system tumors [2], GS is regarded as a distinct variant of glioblastoma multiforme (GBM). The mesenchymal component in GSs appears to harbor cytogenetic and molecular abnormalities similar to those found in GSs’ glial component. GSs are genetically similar to primary GBMs in that 20–40% of both tumor entities harbor TP53 mutations, PTEN deletions, and CDKN2A deletions. The exception to this genetic similarity is the relative infrequency of EGFR amplification in GSs [3–5]. GSs represent less than 5% of all glioblastomas and are similar to GBM in having a slight male predominance and affecting patients in all age groups, with an increasing incidence in the elderly [6]. In most cases, GSs are located supratentorially, with a predilection for the temporal lobe, followed by the frontal, parietal, and occipital lobes [1, 7]. As in GBM, the prognosis in GS is poor [1, 6, 8].

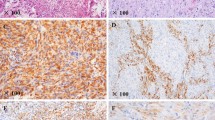
Histology of patient no. 2. a Astroglial component with increased mitotic activity and necrosis, hematoxylin and eosin staining; b sarcomatous component with spindle cells, hematoxylin, and eosin staining; c expression of glial fibrillary acidic protein (GFAP) in subpopulations of the astroglial tumor component, immunohistochemistry; d reticulin fibers are restricted to vessels in the astroglial component (left), but very dense in the sarcomatous component (right), silver staining. By immunohistochemistry, no nuclear accumulation of p53 was demonstrated
Very few reports on pediatric GS are available in the literature. To the best of our knowledge, only 19 pediatric cases of GS with individual data have been published in the English literature to date [9–24]. In the current report, we retrospectively reviewed the data for four pediatric patients with GS who had been enrolled in the HIT-GBM trials in Germany, Austria, and Switzerland since 1994 and summarized data for the 19 cases from the published literature. The 23 pediatric GS cases reviewed herein represent the largest series of this rare tumor in children to date.
Patients and methods
Patient characteristics and inclusion criteria
We searched the HIT-GBM database of the Society of Pediatric Oncology and Hematology in Germany, Austria, and Switzerland (Gesellschaft fuer Paediatrische Onkologie und Haematologie; GPOH) to identify pediatric cases of GS. In accordance with the Declaration of Helsinki, all patients whose information is in this database and/or their parents had given informed consent for statistical analyses and data storage at the time of enrollment in the various HIT-GBM trials. Inclusion criteria for this retrospective review were: (1) a histopathologic diagnosis of GS confirmed by central neuropathologic review (performed by pathologists in the German Brain Tumor Reference Center, Department of Neuropathology, Bonn, Germany) and (2) a patient age of 0–21 years at the time of initial diagnosis.
Statistical analysis
The identified cases from the HIT-GBM database and from the literature were all used for the statistical analysis. Statistical analysis was performed using the statistical package for social studies (SPSS® Inc, Chicago, IL). Overall survival (OS) and event-free survival (EFS) were determined by Kaplan–Meier analysis and log-rank testing. An event was defined as tumor relapse or progression, occurrence of a secondary malignancy, or death from any cause. Patients were grouped according to these parameters: sex (male/female), age (<3 years/≥3 years), preceding radiotherapy of the brain (yes/no), and extent of tumor resection (gross total tumor resection defined as 100% macroscopic removal of tumor mass versus non-total tumor resection). We compared these parameters for prognostic relevance to EFS and OS. For all statistical analyses, significance was set at P < 0.05.
Case reports and summaries
Our search of the HIT-GBM database identified four GS patients who met our criteria and whose data we retrospectively reviewed. The literature search identified 19 GS cases whose data we summarized.
Patient no. 1
In June 2000, the 8-year-old girl was admitted to hospital with generalized seizures and a 2-month history of headache. The MRI showed a brain tumor localized in the right temporal lobe, and the patient underwent a craniotomy with partial resection of the tumor. The histological diagnosis of GS was based on the heterogeneous picture with components of pleomorphic glial cells and a high amount of collagen fibers besides a pleomorphic component with reticulin fibers and leukocyte infiltration. Both histological components expressed p53 and MIB-1 (5–10%) and less intense S100 and GFAP. Since a pleomorphic xanthoastrocytoma was taken into consideration as the underlying diagnosis, no adjuvant therapy was given until the tumor showed further progression 2 months later. At this time gross total resection was achieved. The patient received combined radiochemotherapy according to the HIT-GBM C protocol [25], but the tumor was again progressive 5 months after initial diagnosis with intracranial and spinal cord metastases. Chemotherapy was modified to carboplatin/etoposide. After one cycle the patient died from pneumonia.
Patient no. 2
The 10-year-old boy was admitted to hospital in March 2005 with generalized seizures and a 4-month history of headache and vomiting. Cranial MRI showed a polycystic tumor (6 × 8 × 6 cm) in the right temporal lobe with contrast enhancement of the solid areas (Fig. 2). The patient underwent a craniotomy with gross total tumor resection. On histology a biphasic histopathological pattern with a sarcomatous as well as a glial component led to the diagnosis of GS. Both tumor parts showed an increased proliferation (MIB-1 index 5%) and no nuclear accumulation of p53. The patient received a combined radiochemotherapy according to the HIT-GBM D protocol beginning with pre-irradiation high-dose methotrexate and continuing with simultaneous radiochemotherapy and consolidation chemotherapy until March 2006 [26]. Due to side effects, some chemotherapy cycles had to be modified. In April 2006, the patient reported paresthesia of the lower limbs, and a spinal MRI showed multiple metastases in the spinal canal. In the following month two different chemotherapy regimens were applied. Initially, chemotherapy with carboplatin/etoposide led to a temporary reduction of pain medication, but chemotherapy was accompanied by a neutropenic fever episode as well as the need for platelet transfusions. Since overall mobility was not improved, and weight loss continued, patient and parents asked for cessation of this chemotherapy and temozolomide was started but was accompanied by distinct nausea, increasing insomnia and marked thrombopenia. After acceptance of the palliative situation by the patient and parents, the decision was made to continue with pure palliative symptom control without further chemotherapy. The patient died at home 18 months after the initial diagnosis.

Brain MRI of patient no. 2 at first presentation. T2 coronal (a) and T1 axial sections (b, with contrast) show a contrast-enhancing solid tumor (TU) with a cystic component (TC) and an extensive peritumoral edema (ED). Both tumor and edema cause a marked midline shift (MS) as a correlate of significantly increased intracranial pressure
Patient no. 3
In April 2005 the 6-year-old boy was admitted to the hospital since he had become unconscious after a head trauma. His mother reported on vomiting for 4 months. The CT showed a large hemorrhage in the right frontal lobe leading to a herniation of the brain stem. Thus, an immediate decompressive craniotomy was performed in which a tumor of the right frontal lobe (6 × 5 × 4 cm) was found and partially resected. The histopathologic diagnosis of GS was based on the biphasic pattern with glial tumor cells showing numerous atypical mitoses and a sarcomatous component. Ki67 staining (MIB-1 index) demonstrated up to 30% positive tumor cells. The patient received combined radiochemotherapy according to the HIT-GBM D protocol with pre-irradiation high-dose methotrexate [26]. Nevertheless, a further local progression and leptomeningeal spread to the spinal cord was found. Since his general condition worsened rapidly, chemotherapy was discontinued, and the patient was discharged for home palliative care.
Patient no. 4
The 9-year-old boy was admitted to the hospital in February 2007 with vomiting, diplopia and vertigo following a suspected head trauma. The cranial MRI showed a tumor of the diencephalon and the right cerebral peduncle. A craniotomy was performed, but the tumor could only be resected partially. On histology a biphasic pattern with sarcomatous and glial components was found, and the diagnosis of GS was made. On immunohistochemistry 30% of the tumor cells overexpressed p53-protein, and up to 10% expressed MIB-1. The patient was enrolled in the HIT-GBM D trial [26] and was treated with simultaneous radiochemotherapy followed by consolidation chemotherapy. After 8 months, the patient showed tumor progression. Temozolomide chemotherapy was started. After 12 months, the patient was admitted to the hospital because clinical deterioration due to tumor progression with extension into both thalamic regions. At the last reported follow-up 14 months after the initial diagnosis, the patient had been discharged home in a still significantly reduced condition. To the best of our knowledge, this is the first pediatric patient with a GS located in the mesencephalon published to date.
A further gliosarcoma patient was identified in the HIT-GBM database in whom the diagnosis was made by both the local neuropathologist and the central review by the German Brain Tumor Reference Center. During the work on this paper, the tumor was reevaluated by an international board of pediatric neuropathologists, and the diagnosis was revised to desmoplastic infantile astrocytoma with pronounced anaplastic features. Although we excluded this patient from the following analysis, we are still reporting this case, since it demonstrates the difficulty of the histological classification of rare pediatric brain tumors and the need for a central review with wide experience.
In this patient, in December 2003, routine cerebral ultrasound scan screening showed a large polycystic structure in the right temporo-parietal lobe of the newborn. During the following weeks the lesion grew rapidly, causing hydrocephalus. A craniotomy and partial resection of the tumor were performed. On histology a biphasic tissue pattern with mainly sarcomatous components but also small glial parts with necrosis were seen. GFAP staining was positive within the glial parts, and the MIB-1 index was as high as 20% in both components. After the diagnosis of GS, the patient was treated according to the HIT SKK protocol [27] with intensive multiagent chemotherapy. After 5 months of chemotherapy, the residual tumor presented with a sharp demarcation on MRI; therefore, a second craniotomy was performed resulting in gross total tumor resection. At the last reported follow-up 25 months after the initial diagnosis, the patient was in good clinical condition with no signs of tumor progression.
Patients nos. 5 to 23
In addition, we identified 19 cases of pediatric GS previously reported in the English literature [9–24]. These cases are summarized in Table 1.
Results
The 23 cases comprised 12 males and 10 females; gender was not reported in one case. The median patient age was 11 years (range 0–21) (Table 1). Figure 3 presents a comparison of age distribution for GS and GBM cases. There was a peak accumulation of GS cases in infants: 6 (26%) of the 23 patients were under 3 years of age at diagnosis (Fig. 3).

Comparison of age distribution for gliosarcoma (GS) and glioblastoma multiforme (GBM) cases. Most of the patients with GBM were adolescents (black line, data from the HIT-GBM database). In contrast, the age distribution of GS patients showed two peaks: in addition to a peak in the teenage years, an additional peak was noted in infants. Of the 23 patients reviewed in our series, 6 were under 3 years of age
Duration of clinical history was short with a median of 0.7 month (range 0–4.0 months). Signs of raised intracranial pressure were the leading initial symptoms with vomiting and/or headache (14/21 cases, 67%) and macrocephaly (mainly infants, 5/21 cases, 24%). Other less frequent symptoms were psychomotoric slowing and/or reduced consciousness in five patients (24%), hemiparesis in four patients (19%) and seizures in three patients (14%).
Secondary gliosarcomas were found in five patients after a previous history of cranial radiotherapy for the following malignancies: low grade glioma, medulloblastoma, acute lymphoblastic leukemia, meningioma and giant cell glioblastoma (Table 1). In addition, one patient had received radiotherapy of the scalp due to an angioma. Thus, in total, 26% (6 out of 23) of all pediatric GS patients had received irradiation of the brain between 29 months and 12 years before GS diagnosis. However, none of the four patients from the HIT-GBM data base developed a GS after previous cranial radiotherapy.
With one exception (patient no. 4), GS was strictly localized in the cerebral hemispheres (96%, 22 out of 23 cases; Table 1) with a predilection of the frontal lobes (11 cases, 48%) followed by both the parietal and temporal lobes (10 cases each, 43%). The occipital lobe was affected in seven cases (30%). The exception was our case no. 4, with the primary tumor in the mesencephalon. GS in other regions of the CNS were only found in case of metastasis (Tables 1 and 2).
Gross total tumor resection was achieved in 60% of GS patients (12 out of 20 cases, extent of surgery was not reported in 3 cases).
The median OS was 12.1 months and median EFS 9.8 months. One-year OS was 55 ± 11%, and 2-year OS was 30 ± 11% (Fig. 4). One-year EFS was 44 ± 11%, and the 2-year EFS was 30 ± 11% (Table 2). We compared different groups in Kaplan–Meier survival analyses. Patients who underwent gross total tumor resection had a survival of 13.3 months versus 12.1 months if gross total tumor resection was not achieved, but the difference was not significant. The apparent inhomogeneous age distribution did not translate into significant differences in survival, although survival was superior in the four patients under 3 years of age at diagnosis versus older patients: Here, 2-year OS was 50 ± 25% versus 21 ± 12%, and 2-year EFS was 50 ± 25% versus 22 ± 13% in older patients. Gender did not have an impact on survival. Patients who developed a secondary GS after previous radiotherapy did not show an inferior outcome when this analysis was restricted to patients older than 3 years of age. Median OS and EFS of this group were 13.3 and 7.0 months versus 12.1 and 9.8 months for non-infant patients without previous radiation (not significant).

Overall survival in 23 pediatric patients with gliosarcoma
Discussion
In 23 pediatric patients with gliosarcoma, we found an inhomogeneous bipolar age distribution with one peak in the first year of age and a second broader peak in teenage patients. The frequency of previous radiotherapy among GS patients from the literature was high, although similar results were not found in the pediatric GS patients from the HIT-GBM database. Survival was as poor as generally reported for GBM.
The relative incidence in our HIT-GBM database was 1.9% GS among pediatric glioblastomas (4 out of 206 cases), which is less than in adult patients where GS accounts for up to 5% of glioblastomas [1, 4, 6]. The gender distribution [1, 6, 8] and the tumor location appear to be comparable between adult and pediatric patients [8, 28]. Most tumors were supratentorially found with a strong predominance of the temporal lobe in most adult series, while in our cohort the frontal lobe was most frequently affected. Signs of increased intracranial pressure (vomiting, headache and macrocephaly) were the leading symptoms of GS in pediatric patients. Psychomotoric slowing and/or reduced consciousness, hemiparesis and seizures represented other less frequent initial symptoms. These findings are in good concordance with a previously published series of 29 mainly adult patients presenting with weakness (50%), headache (40%) and seizures (10%) [7]. Probably due to the superficial localization, gross total tumor resection was feasible in as many as 60% (12 out of 20 patients; in 3 missing data), but in contrast to a previously published series of adult gliosarcoma patients [6] the survival benefit from gross total resection did not reach significance (data not shown) [29–31].
With all these characteristics so similar between the pediatric and the adult GS population, and between the pediatric GS and pediatric GBM population, the two-peak age distribution is even more surprising. Holt described the first case of an infant with GS in 1917 [32], but since then relatively little has become known about this peculiar entity. Four of the patients in our series were young infants (1–4 months of age at diagnosis), and the oldest of them showed an increased head circumference at birth [22]. These patients may represent congenital GS. The survival of the young patients in our series was surprisingly good, in particular when considering that radiation cannot play the same predominant role in this population as in older patients. This is in keeping with the experience of other reports [33–36], and it supports the hypothesis that infant GS might represent a disease quite distinct from typical GBM. The prognosis of those patients is good, and the tumors should be resected whenever possible.
A second peculiar group of patients was identified in the literature review: six of the pediatric patients developed a secondary GS after previous radiotherapy for another disease: medulloblastoma, giant cell glioblastoma, low grade glioma, meningioma, acute lymphatic leukemia and angioma. This parallels the experience in adult patients reported by others: Perry and coworkers reported on 32 patients with GS, out of whom 7 had previous irradiation for glioblastoma multiforme [37]. However, in Perry's study a counterintuitive better survival was found in those patients with secondary GS, which was not true in our patients: There was no significant difference in OS and EFS in GS with or without previous cranial radiotherapy.
In conclusion, the present study encompassed the largest cohort of pediatric patients with GS to date. It shows that GS represented a rare tumor entity in children with a relative incidence of 1.9% among all GBM, and hence, epidemiologic and prognostic analysis is only feasible by including previously published cases from the literature. Therefore, interpretation of data is limited to its retrospective character. Even this rare group could be separated into three distinct populations. The tumor is relatively frequent in infants and very young children, and has a particularly good prognosis in this group even when treated only with resection and chemotherapy without radiation, so efforts should be undertaken to completely resect these tumors. A second population of patients developed GS as secondary malignancy after cranial radiotherapy. These patients were older, but in contrast to adults, the survival was as poor as in the third population who developed GS de novo. These patients shared the particularly poor prognosis with adult GS patients or other GBM patients. In the future, molecular pathological analyses might corroborate our observations and further elucidate the biology of this rare tumor entity in children.
References
Ohgaki H, Biernat W, Reis R, Hegi M, Kleihues P (2000) Gliosarcoma. In: Kleihues P, Cavenee WK (eds) Pathology and genetics of tumours of the nervous system. IARC Press, Lyon, pp 42–44
Kleihues P, Burger PC, Aldape KD (2007) Glioblastoma. In: Louis DN, Ohgaki H, Wiestler O, Cavenee WK (eds) WHO classification of tumors of the central nervous system. IARC Press, Lyon, pp 33–49
Kleihues P, Sobin LH (2000) World Health Organization classification of tumors. Cancer 88:2887
Miller CR, Perry A (2007) Glioblastoma. Arch Pathol Lab Med 131:397–406
Reis RM, Konu-Lebleblicioglu D, Lopes JM, Kleihues P, Ohgaki H (2000) Genetic profile of gliosarcomas. Am J Pathol 156:425–432
Kozak KR, Mahadevan A, Moody JS (2009) Adult gliosarcoma: epidemiology, natural history and factors associated with outcome. Neurooncology 11(2):183–191
Sarkar C, Sharma MC, Sudha K, Gaikwad S, Varma A (1997) A clinico-pathological study of 29 cases of gliosarcoma with special reference to two unique variants. Indian J Med Res 106:229–235
Lutterbach J, Guttenberger R, Pagenstecher A (2001) Gliosarcoma: a clinical study. Radiother Oncol 61:57–64
Salvati M, Lenzi J, Brogna C, Frati A, Piccirilli M, Giangaspero F, Raco A (2006) Childhood’s gliosarcomas: pathological and therapeutical considerations on three cases and critical review of the literature. Childs Nerv Syst 22:1301–1306
Radkowski MA, Naidich TP, Tomita T, Byrd SE, McLone DG (1988) Neonatal brain tumors: CT and MR findings. J Comput Assist Tomogr 12:10–20
Ono N, Nakamura M, Inoue HK, Tamura M, Murata M (1990) Congenital gliosarcoma; so-called sarcoglioma. Childs Nerv Syst 6:416–420
Okami N, Kawamata T, Kubo O, Yamane F, Kawamura H, Hori T (2002) Infantile gliosarcoma: a case and a review of the literature. Childs Nerv Syst 18:351–355
Malde R, Jalali R, Muzumdar D, Shet T, Kurkure P (2004) Gliosarcoma occurring 8 years after treatment for a medulloblastoma. Childs Nerv Syst 20:243–246
Kaschten B, Flandroy P, Reznik M, Hainaut H, Stevenaert A (1995) Radiation-induced gliosarcoma. Case report and review of the literature. J Neurosurg 83:154–162
Cerame MA, Guthikonda M, Kohli CM (1985) Extraneural metastases in gliosarcoma: a case report and review of the literature. Neurosurgery 17:413–418
Rizk T, Nabbout R, Koussa S, Akatcherian C (2000) Congenital brain tumor in a neonate conceived by in vitro fertilization. Childs Nerv Syst 16:501–502
Lach M, Wallace CJ, Krcek J, Curry B (1996) Radiation-associated gliosarcoma. Can Assoc Radiol J 47:209–212
Lee YY, Castillo M, Nauert C, Moser RP (1985) Computed tomography of gliosarcoma. AJNR Am J Neuroradiol 6:527–531
Deb P, Sharma MC, Chander B, Mahapatra AK, Sarkar C (2006) Giant cell glioblastoma multiforme: report of a case with prolonged survival and transformation to gliosarcoma. Childs Nerv Syst 22:314–319
Takaue Y, Sullivan MP, Ramirez I, Cleary KR, van Eys J (1986) Second malignant neoplasm in treated Hodgkin’s disease. Report of a patient and scope of the problem. Am J Dis Child 140:49–51
Chadduck WM, Gollin SM, Gray BA, Norris JS, Araoz CA, Tryka AF (1987) Gliosarcoma with chromosome abnormalities in a neonate exposed to heptachlor. Neurosurgery 21:557–559
Goldstein SJ, Young B, Markesberry WR (1981) Congenital malignant gliosarcoma. AJNR Am J Neuroradiol 2:475–476
Kepes JJ, Bastian FO, Weber ED (1996) Gliosarcoma developing from an irradiated ependymoma. Acta Neuropathol (Berl) 92:515–519
McKeever PE, Wichman A, Chronwall B, Thomas C, Howard R (1984) Sarcoma arising from a gliosarcoma. South Med J 77:1027–1032
Wagner S, Leuthold U, Schmid H-J, Wolff JE (2003) Pilotstudie mit hochdosiertem Methotrexat und nachfolgender simultaner Radiochemotherapie bei neun Kindern mit hochgradig-malignen Gliomen (abstract). Monatschr Kinderheilkd 151:467–476
Wagner S, Reinert C, Schmid HJ, Liebeskind AK, Jorch N, Langler A, Graf N, Warmuth-Metz M, Pietsch T, Peters O, Wolff JE (2005) High-dose methotrexate prior to simultaneous radiochemotherapy in children with malignant high-grade gliomas. Anticancer Res 25:2583–2587
Rutkowski S, Bode U, Deinlein F, Ottensmeier H, Warmuth-Metz M, Soerensen N, Graf N, Emser A, Pietsch T, Wolff JE, Kortmann RD, Kuehl J (2005) Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. N Engl J Med 352:978–986
Morantz RA, Feigin I, Ransohoff J 3rd (1976) Clinical and pathological study of 24 cases of gliosarcoma. J Neurosurg 45:398–408
Kramm CM, Wagner S, Van Gool S, Schmid H, Strater R, Gnekow A, Rutkowski S, Wolff JE (2006) Improved survival after gross total resection of malignant gliomas in pediatric patients from the HIT-GBM studies. Anticancer Res 26:3773–3779
Heideman RL, Kuttesch J Jr, Gajjar AJ, Walter AW, Jenkins JJ, Li Y, Sanford RA, Kun LE (1997) Supratentorial malignant gliomas in childhood: a single institution perspective. Cancer 80:497–504
Artico M, Cervoni L, Celli P, Salvati M, Palma L (1993) Supratentorial glioblastoma in children: a series of 27 surgically treated cases. Childs Nerv Syst 9:7–9
Holt LE (1917) Gliosarcoma in an infant of seven weeks, resembling hydrocephalus. Am J Dis Child 14:219–221
Duffner PK, Horowitz ME, Krischer JP, Friedman HS, Burger PC, Cohen ME, Sanford RA, Mulhern RK, James HE, Freeman CR et al (1993) Postoperative chemotherapy and delayed radiation in children less than three years of age with malignant brain tumors. N Engl J Med 328:1725–1731
Dufour C, Grill J, Lellouch-Tubiana A, Puget S, Chastagner P, Frappaz D, Doz F, Pichon F, Plantaz D, Gentet JC, Raquin MA, Kalifa C (2006) High-grade glioma in children under 5 years of age: a chemotherapy only approach with the BBSFOP protocol. Eur J Cancer 42:2939–2945
Geyer JR, Finlay JL, Boyett JM, Wisoff J, Yates A, Mao L, Packer RJ (1995) Survival of infants with malignant astrocytomas. A Report from the Childrens Cancer Group. Cancer 75:1045–1050
Wagner S, Rutkowski S, Reinert C, Straeter R, Slavc I, Pietsch T, Van Gool S, Wolff JE (2006) High grade gliomas in infants and very young children: a retrospective analysis of 30 patients (abstract). Pediatr Blood Cancer 47:356–532
Perry JR, Ang LC, Bilbao JM, Muller PJ (1995) Clinicopathologic features of primary and postirradiation cerebral gliosarcoma. Cancer 75:2910–2918
Acknowledgments
The ongoing support of the Deutsche Kinderkrebsstiftung, Bonn, Germany, is greatly acknowledged. Without this support performance of clinical trials as well as quality control measures like central neuropathological and neuroradiological review and a central review of radiotherapy planning would not be possible within the HIT network. We also thank all colleagues who contributed patients and their data to the HIT-GBM studies.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Karremann, M., Rausche, U., Fleischhack, G. et al. Clinical and epidemiological characteristics of pediatric gliosarcomas. J Neurooncol 97, 257–265 (2010). https://doi.org/10.1007/s11060-009-0021-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-009-0021-3