
Abstract
Several recent studies have shown that aberrant constitutive activation of nuclear factor kappaB (NF-κB) is present in a variety of cancers including gliomas. NF-κB is known to play important roles in the physiological regulation of diverse cellular processes such as inflammation, growth and immunity. In contrast, aberrant activation of this latent transcription factor promotes cancer cell migration, invasion and resistance to chemotherapy. Here we show by electro-mobility shift assay (EMSA) and immuno-staining that constitutive NF-κB activation is present in various malignant glioma cell lines as well as in primary cultures derived from tumor tissue. This activation was not serum dependent and it led to high IL-8 gene transcription and protein production. Over-expression of an I-κB super-repressor (I-κB SR) transgene completely blocked constitutive NF-κB activation, nuclear localization and transcription of some but not all NF-κB regulated genes indicating that NF-κB signaling in glioma cells is I-κB dependent. Surprisingly, over-expression of IκBSR did not have any effect on the transcription levels of anti-apoptotic genes in these glioma cultures and cell lines. Down-regulation of NF-κB activation reduced invasion of glioma cells through matrigel. Collectively these data suggest that aberrant constitutive activation of NF-κB in glioblastoma cells promotes their invasive phenotype. Interruption of this aberrant NF-κB activity may help reduce the spread of this infiltrative tumor.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glioblastoma Multiforme (GBM) is a devastating disease, which accounts for 40% of all primary malignant tumors of the nervous system. Despite the use of surgery, cytotoxic chemotherapy and radiotherapy, median survival of these patients is only about 1 year. GBM tumors have two distinct components: an enhancing mass with distinct borders and a non-enhancing infiltrative region, which may extend many centimeters from the enhancing mass. Surgical resection of the enhancing mass can provide modest survival benefit, but is not curative as the infiltrative component of the tumor usually resides in functioning cortex and cannot be removed safely [1]. Non-surgical therapies including cytotoxic chemotherapy and radiotherapy are used to treat the infiltrative component of the tumor, but also are not curative. An improved understanding of the biology of GBM tumor cell infiltration may help to guide the development of new therapeutic approaches for treating this deadly disease.
Nuclear factor kappaB (NF-κB) is a transcription factor consisting of Rel family proteins. When activated, these proteins form heterodimers, most commonly containing P65 and P50 proteins. In quiescent cells, NF-κB remains in the cytosol bound to its inhibitory component IκB. Activation of NF-κB occurs when IκB gets phosphorylated, ubiquitinated and subsequently degraded. NF-κB is then free to translocate to the nucleus where it binds to the cognate binding sites of the promoters of multiple genes which are involved in a variety of processes including proliferation and survival [2]. Several studies have also suggested that this transcription factor plays an important role in the regulation of genes involved in cellular adhesion, migration and invasion [3–10].
Activation of NF-κB is a double-edged sword, as it is needed for proper immune function but inappropriate activation leads to abnormal cell proliferation, inflammation, tumorigenesis or metastasis [11]. NF-κB also plays an important role in regulating tumor cell infiltration as it is required for the expression of many adhesion molecules [12] and matrix-metallo proteases (MMP) that are responsible for invasion [13]. NF-κB is known to be constitutively activated in many types of cancer [14–19]. There is some evidence that gliomas also have aberrantly activated NF-κB [20–22].
Here we show that NF-κB is aberrantly constitutively activated in glioma cell lines and also in primary cell cultures established from tumors obtained directly from the operating room. Surprisingly, interruption of NF-κB activation did not affect proliferation or anti-apoptotic gene expression in GBM cell lines or cultures but it did reduce glioma cell invasion.
Materials and methods
Reagents
Dulbecco’s modified Eagle’s medium (DMEM) and fetal Bovine serum (FBS) were obtained from GIBCO-BRL (Gathersburg, MD). All antibodies were obtained from Santa Cruz Biotechnology.
Cell cultures
GBM cell lines D54, U251, LN-229 and U87, and primary cultures from clinically harvested glioblastoma tumors were grown in DMEM supplemented with 10% FBS and penicillin.
Primary cell cultures established from human GBMs obtained from surgery were minced in Petri dishes containing DMEM and grown in serum-free medium. Once clones were established, the cells were expanded in DMEM supplemented with 10% fetal Bovine serum (FBS). Each clone was verified to consist of tumor cells by analysis of loss of heterozygosity (LOH) of chromosome 10, which is present in about 80% of GBMs [23]. Only tumor cultures which shared LOH with the parental tumors were considered to be free of non-neoplastic contaminants.
Preparation of whole-cell extracts
Protein extracts were prepared from cell cultures grown in monolayer in DMEM in a tissue culture incubator at 37°C and 5% CO2. Cells were resuspended in extraction buffer (20 mM Tris, pH 8.0;150 mM MgCl2; 1% Triton ×100) containing protease inhibitor cocktail, and were kept on ice for 20 min. The crude extracts were centrifuged at 18,000 × g for 20 min at 4°C to clear debris, and supernatants representing whole-cell extracts (WCE) were collected. WCEs were aliquoted in small volumes in order to minimize repeated freeze-thaw damage, and were kept at −80°C for further use. The protein content of WCEs was determined with the bicinchoninic acid (BCA) protein assay (Pierce, Rockford, IL).
Electrophoretic mobility shift assay
For electrophoretic mobility shift assay (EMSA), 10 μg of the WCEs were incubated in binding buffer (8 mM N-2-hydroxyethylpiperazine-N′-2-ethane sulfonic acid, pH 7.0; 10% glycerol; 20 mM KCl; 4 mM MgCl2; 1 mM sodium pyrophosphate) containing 1.0 μg of polydeoxyinosine-deoxycytosine and 32P end-labeled probes. The probes had the following sequences: 5′-AACTCCGGGAATTTCCCTGGCCC-3′; 5′-GGGCCAGGGAAATTCCCGGAGTT-3′. For competition experiments, a 1,000-fold excess of cold oligonucleotide was used. For the supershift assay each WCE was incubated with anti-p65 or anti-p50 antibody (Santa Cruz Biotechnology, Santa Cruz, CA) for 30 min at room temperature (RT) before the addition of probe. After incubation with the probe for 20 min, the reaction mixture was analyzed on a 4% non-denaturing acrylamide gel. The gels were then dried and exposed for autoradiography.
Luciferase assay
For Luciferase assay we used a κB-luciferase plasmid (p5XIP10κB), which contains five tandem copies of the NFκB site from the IP10 gene, driving the luciferase gene [24]. Luciferase assay was performed using Renilla Luciferase Assay system from Promega according to the manufacturer’s protocol.
Viral gene transfer
Inactivation of NF-κB was performed using a replication deficient adenovirus containing a transgene coding for the irreversible repressor of IκB-α (IκB super-repressor (IκBSR), obtained from Lerner Research Institute virus core). An empty adenovirus was used as vector control (Ad-ΔE). Recombinant adenovirus expressing the green fluorescence protein (GFP) gene was used to standardize experimental conditions. Cells were transduced at multiplicities of infection (MOI) of 20 in serum free DMEM for 1 h with gentle shaking and then washed with phosphate buffered saline (PBS) then incubated with growth medium at 37°C, 5% CO2 until use. Cells were harvested at various time points according to the experimental plan.
RNAse protection assay
RNA was prepared using the RNA easy protocol (Qiagen, Valencia, CA). RNase protection assay was performed using a PharMingen kit (BD PharMingen). L32 and GAPDH were used as internal controls. Quantification of the radioactivity was done using a StormImager and calculated using ImageQuant software (Molecular Dynamics Inc.).
Matrigel invasion assay
Invasive activity of glioblastoma cells was measured with the use of a modified Boyden chamber containing Matrigel (BD Biosciences, USA). Each chamber includes a well with a membrane containing 8 μM pores. Membranes are coated with a basement membrane matrix (Matrigel). Approximately 2.5 × 104 cells in 500 μl serum free media were seeded on top of the Matrigel. The bottom well contained regular media with serum. Cells were allowed to grow in a tissue culture incubator for 24 h. Cells that remained on the top of the membrane were scrubbed off with a cotton swab. Cells that invaded into the matrix were fixed and stained with hematoxylin blue. At a pre-determined time point, the media was removed, cells that did not invade were scrubbed off, fixed and were stained and counted using the ImageQuant software.
Results
NF-κB is constitutively activated in GBM cell lines and primary tumor cell cultures
Figure 1A shows the EMSA analysis using WCEs prepared from either GBM cell lines (Lanes 2–6) or low passage primary GBM tumor cell cultures (Lanes 7–9). 293 cell extract (Lane 1) was used as a negative control. Lanes 10–13 show the NF-κB activation levels of GBM cell lines maintained in serum-free conditions for 24 h. NF-κB was found to be activated in all cell lines tested. The magnitude of activation varied widely between cell lines and primary cell cultures, but there was no apparent effect of exposure to serum-free culture conditions on the level of constitutive activation. Normal astrocytes do not show constitutively activated NF-κB (data not shown).

Constitutive NF-κB activity of glioblastoma cell lines and primary cultures. (A) EMSAs performed with a NF-κB probe on whole cell extracts of glioblastoma cell lines (lanes 2–6) and primary cultures (lanes 7–9) obtained directly from tumor tissue. Lane 1 shows 293 cells, which do not have detectable NF-κB activity (negative control). Lanes 10–13 show NF-κB activity in serum deprived conditions (24 h). (B) Supershift of the NF-κB band with p50 (Lane 2) and p65 (Lane 3) antibodies in U87 cells (lanes 1–4) and TNF-α treated 293 cells (lanes 5–8, positive control). C) Competition assay in U87 cells. Lanes 2 and 3 show disappearance of binding of the NF-κB probe with the addition of increasing concentrations of the wild-type κB binding site as competitor. Lanes 4 and 5 show that a mutant κB probe failed to compete with binding of the full NF-κB probe
In Fig. 1B, we show that P50 and P65 antibodies produced super shifting of the NF-κB band thereby confirming the presence of a p65/p50 heterodimer in U87 cells. 293 cells treated with TNF-α were used as a positive control for this experiment.
Competition assays with wild type and mutated κb binding sites with 5- and 25-fold molar excess are shown in Fig. 1C. As expected, NF-κB binding was competed out with wild type (lanes 2 and 3) but not with mutated oligos (lanes 4 and 5).
Over-expression of IκB super repressor (IκBSR) inactivates NFκB
GBM cell lines and primary cell cultures were infected with an adenoviral construct containing IκBSR gene or empty vector control with varying MOI and harvested after 24 h. WCEs were prepared and subjected to western blot analysis. A monoclonal antibody to IκB-α (Clone H4, Santa Cruz Biotechnology) was used to detect both the endogenous and the exogenous IκB (Fig. 2A). Increased exogenous expression of IκB protein was seen with increased titers of the virus. EMSA was performed to determine the ability of IκBSR to inactivate NF-κB in glioma cell lines and primary tumor cell cultures. Figure 2B shows that over-expression of I-κBSR inhibited endogenous as well as TNF-α-induced NF-κB activation in both a GBM cell line (D54) and a primary tumor cell culture (CCF-52). These results are representative of all of the cell lines and primary cell cultures examined; in no case did IκBSR over-expression fail to inactivate NF-κB.

Adenoviral IκBSR infection increases I-κB protein expression in glioblastoma cells.(A) GBM cells were infected with 50 and 100 MOI of adenoviral I-κBSR and harvested after 72 h to study the expression of I-κB, using antibody clone H4, Santa Cruz Biotechnology. All three glioblastoma cell lines show considerable expression of the I-κBSR transgene although expression was higher in D54 and U87 than in U251 cells. (B) EMSA was performed to study the effect of I-κBSR over expression on endogenous and TNF-α induced NF-κB DNA binding activity. D54 and CCF-52 cells were infected with an empty adenoviral vector (V) or with the I-κBSR transgene (SR). Cell extracts were prepared after 24 h and DNA binding was examined by EMSA in presence and absence of TNF-α
Down regulation of NF-κB dependent gene transactivation by IκBSR
We used a NF-κB driven reporter to confirm the suppressive effect of IκBSR on the constitutive levels of NF-κB activity. In Fig. 3 we show that NF-κB dependent luciferase activity was reduced following over-expression of I-κBSR in U87 cells. Similar results (activation of the NF-κB reporter with TNF-α, inactivation with I-κBSR) were observed in all GBM cell lines (data not shown).

Use of an NF-κB promoter driven luciferase reporter to examine the effects of TNF-α treatment and I-κBSR over-expression. Stable clone of U87 cells carrying a NF-κB promoter driven luciferase reporter was either untreated (UT) or infected with an empty adenoviral vector (Vec) or an adenovirus carrying the IκBSR transgene (SR). Luciferase activity was measured using a luminometer. Over-expression of IκBSR produced a significant reduction in NFκB dependent gene transcription (t-test, P values as shown)
We next evaluated whether down regulation of NF-κB would affect endogenous gene expression in glioma cell lines. Figure 4A shows the constitutive presence of IL-8 transcripts in almost all established GBM cell lines, except U251, and also in our primary glioma cell cultures. Figure 4B shows that with I-κBSR over-expression, NF-κB binding (top panel, EMSA) is decreased compared with untreated (−) and vector infected (Vec) cell which also corresponds to the steady state IL-8 RNA transcription levels (middle panel, RPA assay) in GBM cell lines and primary cultures. The D54 and U87 cell lines showed complete elimination of IL-8 transcripts upon I-κBSR infection. Figure 5 shows that IκBSR over-expression also reduced IL-8 protein production compared to untreated controls in U87 (left panel) and LN-229 (right panel) cells. Although we observed a trend towards reduced IL-8 production in untreated cells over time, this trend was not statistically significant (t-test, P = 0.07–0.21).

I-κBSR over-expression decreases NF-κB DNA binding and results in decreased transcription of IL-8 RNA. (A) Total cellular RNA of the cell lines and primary cultures listed were extracted and subjected to RNAse protection assay for IL-8 transcripts. L32 and GAPDH were used as internal control. (B) CCF4, CCF52, D54 and U87 cells were infected either with an empty adenovirus vector (Vec) or adenovirus containing the I-κBSR (SR) transgene. Top panel shows NF-κB binding (by EMSA); the lower panels show IL-8 and L32 (control) transcripts measured by RPA

I-κBSR over-expression decreases IL-8 protein production by GBM cells. IL-8 protein production by U87 (left) and LN229 (right) cells was determined by ELISA assay. I-κBSR over-expression (cross-hatched bars) decreased IL-8 protein production significantly (*) in both cell lines compared to uninfected control (solid bars). Although the untreated cells showed a trend towards lower IL-8 production over time, this trend did not reach statistical significance (P = 0.07−0.26)
Downregulation of NF-κB does not affect pro- or anti-apoptotic gene transcription
The effect of NF-κB inhibition on the expression of members of the Bcl-2 gene family including Bcl-xL, BID, Bax and Mcl-1 was examined by RPA in GBM cells following 24 h treatment with media, empty adenoviral vector or adenovirus containing the I-κBSR construct. L-32 and GAPDH were used as internal controls for quantitation of the RPA results. Successful over-expression of I-κBSR and down-regulation of NF-κB was confirmed by EMSA. Surprisingly, down regulation of NF-κB had little effect on the transcription levels of these Bcl-2 family genes in the glioma cell lines and primary cultures studied (Fig. 6). We also found that down regulation of NF-κB by over-expression of I-κBSR did not produce apoptosis (data not shown).

I-κBSR over-expression does not affect transcription of members of the bcl-2 gene family. CCF52, D54, CCF4 and U87 cells were left untreated (−) or infected either with empty adenovirus vector (Vec) or containing the IκBSR transgene (SR) for 24 h and then RNA was collected for RNAse protection assay. Over-expression of IκBSR did not alter the transcription of bcl-2 family members in any of the cell lines examined
Down regulation of NF-κB activity suppresses GBM cell invasion
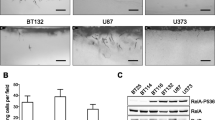
We examined the ability of I-κBSR over-expression to alter the invasive properties of U87 and D54 GBM cells. We used a modified Boyden chamber containing matrigel to perform the invasion assay. Briefly, 2.5 × 104 uninfected, adenoviral vector-infected and adenoviral IκBSR infected U87 cells were seeded in the top well and allowed to invade through matrigel for 24 h. The bottom well contained complete medium. After 24 h, any remaining cells from the upper well were scrubbed off and cells that had invaded through matrigel and underlying membrane were fixed, stained, visualized under 100× magnification and photographed (Nikon Digital camera DXM1200F, Fig. 7A left panel). Imagepro software (Molecular Dynamics) was used to count cells (all cells were counted). We found that over-expression of IκBSR significantly (P = 0.04) reduced the invasiveness of U87 cells compared to vector-only infected cells (Fig. 7A, photomicrographs). Similar to what we observed in U87 cells, we found that over-expression of IκBSR in D54 cells resulted in less invasion when compared to untreated and vector-infected cells. Photomicrographs show a representative example of three separate experiments (Fig. 7B).

Inhibition of NF-κB activity in GBM cells results in reduction of invasion through matrigel. (A) Photomicrographs (left): U87 cells were infected with empty adenoviral vector or with an adenovirus carrying IκB-SR transgene for 24 h. Cells were then counted and 2.5 × 104 cells in 500 μl of serum free media were plated on a matrigel coated well of a modified Boyden chamber. At 24 h after plating, cells on the underside of the membrane were stained and visualized. Right: Graphical representation of the results of 3 separate experiments. (B) Photomicrographs showing invasion assay as in A, with use of D54 cells. These are representative images of 3 experiments
Super-activation of NF-κB activity increases GBM cell invasion
We examined the effects of super-activation of NF-κB on the invasive phenotype of glioma cell lines. We treated the cells with TNF-α in order to super-activate NF-κB. For the evaluation of invasion, cells were allowed to invade in the presence or absence of TNF-α. After 24 h non-invasive cells were removed from the upper chamber by scrubbing with a cotton swab and the invasive cells were stained with hematoxylin and counted. Figure 8A (left panel) shows an example of the results of this assay (representative pictures from 3 separate experiments). We found that TNF-α treatment increased invasion of U87 cells significantly when compared with untreated controls (P = 0.03) (cell counts shown in Fig. 8, right panel).

Super-activation of NF-κB in U87 cells by TNF-α treatment increases invasion through matrigel. U87 cells were either left untreated (UT) or treated with TNF-α and invasion through matrigel was examined. Left panel shows representative pictures from 3 separate experiments. Right panel shows the graphical representation of the number of cells, which invaded under each treatment condition. TNF-α treated cells showed significantly increased invasion when compared to untreated (UT) cells (P = 0.03)
Discussion
We undertook the current study to more completely investigate the activation status of NF-κB in a panel of GBM cell lines and in primary GBM tumor cell cultures. We also sought to determine the impact of this aberrant activation on the biology of GBM cells with respect to cellular proliferation, apoptosis and invasion.
We found that NF-κB is aberrantly constitutively activated in both GBM cell lines and primary cell cultures. This aberrant NF-κB activity drives gene expression but specific suppression of its activity had no effect on the expression of anti-apoptotic genes or cell proliferation. We did find that the level of NF-κB activity in individual cell lines affected their ability to invade through matrigel. We also observed that suppression of NF-κB activation was associated with decreased IL-8 gene transcription but it had no effect on TGF-beta gene expression (data not shown). Therefore, our results provide evidence for the involvement of NF-κB in the regulation of invasion, but that NF-κB activity did not affect proliferation or apoptosis in GBM cells.
NF-κB is constitutively activated in many cancers of hematopoetic and non-hematopoetic origin. In normal tissues it exists as a latent transcription factor, rarely activated except in cells involved in the regulation of the immune system [11]. We found not only that this transcription factor is constitutively activated but that the level of activation varies amongst cell lines and primary GBM cell cultures (Fig. 1). Aberrant constitutive activation of NF-κB was also present under serum free condition (Fig. 1, lanes 10–13). This finding may indicate that NF-κB activation is maintained via an autocrine mechanism, or that there may be another form of aberrant activation of the upstream signaling pathway. Recently it has been shown that NF-κB can be constitutively activated in an autocrine manner by secreted cytokines [25–28]. Work is continuing in our laboratory to identify the source(s) of NF-κB in our cell lines and primary tumor cell cultures.
We used the transgene IκBSR to suppress both endogenous and TNF-α induced NF-κB activation in GBM cells. Suppression of NF-κB was demonstrated using two separate techniques: EMSA and an in vivo NF-κB dependent luciferase reporter assay. Suppression of NF-κB activation resulted in decreased IL-8 transcription and reduced cellular invasion but no effect on cellular proliferation and transcription of anti-apoptotic genes.
NF-κB is a pleotropic transcription factor that controls the expression of many genes including inducible and constitutive IL-8. The IL-8 promoter has binding sites for several important transcription factors such as activator protein-1 (AP-1), NF-κB and CAAT enhancer binding protein (CEBP) [29]. The significant decrease in NF-κB promoter activity in IκBSR infected cells and down regulation of IL-8 transcripts under those condition strongly suggests that NF-κB tightly regulates IL-8 gene expression in these cells. This finding is consistent with the view that NF-κB promoter is active and indispensable for IL-8 expression in all cell types, including GBM tumor cells [30–31].
Tumor invasion is a complex biological process involving adhesion, motility and degradation of extracellular matrices [32]. Many of the genes regulated by NF-κB are known to be involved in invasion, including MMPs and urokinase type plasminogen activators (uPA) [33]. One of the hallmark characteristics of GBM is infiltrative growth angiogenesis and tumor surveillance.
Certain chemokines, such as IL-8, and growth factors, such as vascular endothelial growth factor (VEGF), are known to play important roles in tumor cell metastasis. IL-8 production is known to be increased in GBM cells and its expression is directly correlated with the histopathological grade of the tumor [34]. Our results show that all GBM cell lines and primary tumor cell cultures expressed constitutive steady state level of IL-8 gene. Over-expression of IκBSR in those cell lines reduced the level of constitutive IL-8 gene expression, which is consistent with the model that NF-κB plays a key role in regulating IL-8 gene expression in GBM cells. Suppression of this important transcription factor in many cancers leads promotion of apoptosis and inhibition of proliferation, metastasis, angiogenesis and invasion [11, 35, 36]. It is not unreasonable to hypothesize that reduced invasion seen in GBM cells over expressing IκBSR is mediated at least in part by IL-8. Indeed, it has been recently reported that treatment of U251 cells with IL-8 increases the invasive potential of this cell line [29]. Furthermore, IL-8 is known to contribute to the invasion of human oral squamous cell [37] through the regulation of MMP-7. In breast cancer cells up-regulation of IL-8 is associated with estrogen receptor inactivation and increased invasion and metastasis [38].
Robe et al. [39] showed that use of the NF-κB inhibitor sulfasalazine and over-expression of IκBSR were able to block cell cycle and induce apoptosis in rat and human glioma cell lines. Surprisingly, we found that down regulation of NF-κB activation did not affect anti-apoptotic gene expression apoptosis or proliferation (data not shown). The reasons underlying these discrepancies are not clear but may relate to the different cell lines and methodologies used.
Ultimately, it will be essential to demonstrate that down regulation of NF-κB results in reduced invasion in an in vivo model of GBM. The effects of pharmacological and genetic manipulations of other signaling pathways on tumor cell invasion have been studied in various isogenic and xenograft animal models of orthotopic GBM [40–42]. We have initiated studies with use of cells that are stably transfected with the I-κBSR transgene, and with pharmacological inhibitors of NF-κB to examine the effects of these manipulations on GBM cells in vivo.
In summary, we have shown that NF-κB is aberrantly constitutively activated in GBM cell lines and primary cell cultures. Down regulation of NF-κB decreases IL-8 gene expression, but not anti-apoptotic members of the Bcl-2 gene family or proliferation it reduces glioma cell invasion. Future studies are planned to more carefully examine the relationship between IL-8 expression and invasion in GBM cells.
References
Sawaya R, Ligon BL, Bindal AK, Bindal RK, Hess KR (1996) Surgical treatment of metastatic brain tumors. J Neurooncol 27:269–277
Baldwin AS Jr (2001) Series introduction: the transcription factor NF-kappaB, human disease. J Clin Invest 107:3–6
Higgins KA, Perez JR, Coleman TA, Dorshkind K, McComas WA, Sarmiento UM, Rosen CA, Narayanan R (1993) Antisense inhibition of the p65 subunit of NF-kappa B blocks tumorigenicity and causes tumor regression. Proc Natl Acad Sci USA 90:9901–9915
Beg AA, Baltimore D (1996) An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science 274:782–784
Gilmore TD, Koedood M, Piffat KA, White DW (1996) Rel/NF-kappaB/IkappaB proteins and cancer. Oncogene 13:1367–1378
Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM (1996) Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science 274:787–789
Wang CY, Mayo MW, Baldwin AS Jr (1996) TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappaB. Science 274:784–787
Duffey DC, Chen Z, Dong G, Ondrey FG, Wolf JS, Brown K, Siebenlist U, Van Waes C (1999) Expression of a dominant-negative mutant inhibitor-kappaBalpha of nuclear factor-kappaB in human head and neck squamous cell carcinoma inhibits survival, proinflammatory cytokine expression, and tumor growth in vivo. Cancer Res 59:3468–3474
Yoshida A, Yoshida S, Ishibashi T, Kuwano M, Inomata H (1999) Suppression of retinal neovascularization by the NF-kappaB inhibitor pyrrolidine dithiocarbamate in mice. Invest Ophthalmol Vis Sci 40:1624–1629
Huang S, DeGuzman A, Bucana CD, Fidler IJ (2000) Nuclear factor-kappaB activity correlates with growth, angiogenesis, and metastasis of human melanoma cells in nude mice. Clin Cancer Res 6:2573–2581
Aggarwal BB Nuclear factor-kappaB: the enemy within (2004) Cancer Cell 6:203–208
Ritchie CK, Giordano A, Khalili K (2000) Integrin involvement in glioblastoma multiforme: possible regulation by NF-kappaB. J Cell Physiol 184:214–221
Bjorklund M, Koivunen E (2005) Gelatinase-mediated migration and invasion of cancer cells. Biochim Biophys Acta 1755:37–69
Griffin JD (2001) Leukemia stem cells and constitutive activation of NK-kappaB. Blood 6:203–208
Feinman R, Koury J, Thames M, Barlogie B, Epstein J, Siegel DS (1999) Role of NF-kappaB in the rescue of multiple myeloma cells from glucocorticoid-induced apoptosis by bcl-2. Blood 93:3044–3052
Kordes U, Krappmann D, Heissmeyer V, Ludwig WD, Scheidereit C (2000) Transcription factor NF-kappaB is constitutively activated in acute lymphoblastic leukemia cells. Leukemia 14:399–402
Baron F, Turhan AG, Giron-Michel J, Azzarone B, Bentires-Alj M, Bours V, Bourhis JH, Chouaib S, Caignard A (2002) Leukemic target susceptibility to natural killer cytotoxicity: relationship with BCR-ABL expression. Blood 99:2107–2113
Palayoor ST, Youmell MY, Calderwood SK, Coleman CN, Price BD (1999) Constitutive activation of IkappaB kinase alpha and NF-kappaB in prostate cancer cells is inhibited by ibuprofen. Oncogene 18:7389–7394
Nakshatri H, Bhat-Nakshatri P, Martin DA, Goulet RJ Jr, Sledge GW Jr (1997) Constitutive activation of NF-kappaB during progression of breast cancer to hormone-independent growth. Mol Cell Biol 17:3629–3639
Bian X, Opipari AW Jr, Ratanaproeksa AB, Boitano AE, Lucas PC, Castle VP (2002) Constitutively active NFkappa B is required for the survival of S-type neuroblastoma. J Biol Chem 277:42144–42150
Nagai S, Washiyama K, Kurimoto M, Takaku A, Endo S, Kumanishi T (2002) Aberrant nuclear factor-kappaB activity and its participation in the growth of human malignant astrocytoma. J Neurosurg 96:909–917
Gill JS, Zhu X, Moore MJ, Lu L, Yaszemski MJ, Windebank AJ (2002) Effects of NFkappaB decoy oligonucleotides released from biodegradable polymer microparticles on a glioblastoma cell line. Biomaterials 23:2773–2781
Hata N, Yoshimoto K, Yokoyama N, Mizoguchi M, Shono T, Guan Y, Tahira T, Kukita Y, Higasa K, Nagata S, Iwaki T, Sasaki T, Hayashi K (2006) Allelic losses of chromosome 10 in glioma tissues detected by quantitative single-strand conformation polymorphism analysis. Clin Chem 52:370–378
Sizemore N, Lerner N, Dombrowski N, Sakurai H, Stark GR (2002) Distinct roles of the Ikappa B kinase alpha and beta subunits in liberating nuclear factor kappa B (NF-kappa B) from Ikappa B and in phosphorylating the p65 subunit of NF-kappa B. J Biol Chem 277:3863–3869
Wolf S, Chen Z, Dong G, Sunwoo JB, Bancroft CC, Capo DE, Yeh NT, Mukaida N, Van Waes C (2001) IL (interleukin)-lalpha promotes nuclear factor-kappaB and AP-1-induced IL-8 expression, cell survival, and proliferation in head and neck squamous cell carcinomas. Clin Cancer Res 7(6):1812–1820
Arlt A, Vorndamm J, Muerkoster S, Yu H, Schmidt WE, Folsch UR, Schafer H (2002) Autocrine production of interleukin 1beta confers constitutive nuclear factor kappaB activity and chemoresistance in pancreatic carcinoma cell lines. Cancer Res 62:910–916
Coward WR, Okayama Y, Sagara H, Wilson SJ, Holgate ST, Church MK (2002) NF-kappa B and TNF-alpha: a positive autocrine loop in human lung mast cells? J Immunol 169:5287–5293
Lu T, Strak G (2004) Cytokine overexpression and constitutive NF-kappa B in cancer. Cell Cycle 3:1114–1117
Wakabayashi K, Kambe F, Cao X, Murakami R, Mitsuyama H, Nagaya T, Saito K, Yoshida J, Seo H (2004) Inhibitory effects of cyclosporin A on calcium mobilization-dependent interleukin-8 expression and invasive potential of human glioblastoma U251MG cells. Oncogene 23:6924–6932
Mahe Y, Mukaida N, Kuno K, Akiyama M, Ikeda N, Matsushima K, Murakami S (1991) Hepatitis B virus X protein transactivates human interleukin-8 gene through acting on nuclear factor kB and CCAAT/enhancer-binding protein-like cis-elements. J Biol Chem 266:13759–13763
Matsusaka T, Fujikawa K, Nishio Y, Mukaida N, Matsushima K, Kishimoto T, Akira S (1993) Transcription factors NF-IL6 and NF-kappa B synergistically activate transcription of the inflammatory cytokines, interleukin 6 and interleukin 8. Proc Natl Acad Sci USA 90:10193–10197
Lefranc F, Brotchi J, Kiss R (2005) Possible future issues in the treatment of glioblastomas: special emphasis on cell migration and the resistance of migrating glioblastoma cells to apoptosis. J Clin Oncol 23:2411–2422
Rao JS (2003) Molecular mechanisms of glioma invasiveness: the role of proteases. Nat Rev Cancer 3:489–501
Brat DJ, Bellail AC, Van Meir EG (2005) The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro-oncol 7:122–133
Huang S, Pettaway CA, Uehara H, Bucana CD, Fidler IJ (2001) Blockade of NF-kappaB activity in human prostate cancer cells is associated with suppression of angiogenesis, invasion, and metastasis. Oncogene 20:4188–4197
Fujioka S, Sclabas GM, Schmidt C, Frederick WA, Dong QG, Abbruzzese JL, Evans DB, Baker C, Chiao PJ (2003) Function of nuclear factor kappaB in pancreatic cancer metastasis. Clin Cancer Res 9:346–354
Watanabe H, Iwase M, Ohashi M, Nagumo M (2002) Role of interleukin-8 secreted from human oral squamous cell carcinoma cell lines. Oral Oncol 38:670–679
Lin Y, Huang R, Chen L, Li S, Shi Q, Jordan C, Huang RP (2004) Identification of interleukin-8 as estrogen receptor-regulated factor involved in breast cancer invasion and angiogenesis by protein arrays. Int J Cancer 109:507–515
Robe PA, Bentires-Alj M, Bonif M, Rogister B, Deprez M, Haddada H, Khac MT, Jolois O, Erkmen K, Merville MP, Black PM, Bours V (2004) In vitro and in vivo activity of the nuclear factor-kappaB inhibitor sulfasalazine in human glioblastomas. Clin Cancer Res 10:5595–5603
Lamszus K, Brockmann MA, Eckerich C, Bohlen P, May C, Mangold U, Fillbrandt R, Westphal M (2005) Inhibition of glioblastoma angiogenesis and invasion by combined treatments directed against vascular endothelial growth factor receptor-2, epidermal growth factor receptor, and vascular endothelial-cadherin. Clin Cancer Res 11(13):4934–4940
Hu B, Jarzynka MJ, Guo P, Imanishi Y, Schlaepfer DD, Cheng SY (2006) Angiopoietin 2 induces glioma cell invasion by stimulating matrix metalloprotease 2 expression through the alphavbeta1 integrin and focal adhesion kinase signaling pathway. Cancer Res 66(2):775–783
Mayes DA, Hu Y, Teng Y, Siegel E, Wu X, Panda K, Tan F, Yung WK, Zhou YH (2006) PAX6 suppresses the invasiveness of glioblastoma cells and the expression of the matrix metalloproteinase-2 gene. Cancer Res 66(20):9809–9817
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Raychaudhuri, B., Han, Y., Lu, T. et al. Aberrant constitutive activation of nuclear factor κB in glioblastoma multiforme drives invasive phenotype. J Neurooncol 85, 39–47 (2007). https://doi.org/10.1007/s11060-007-9390-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-007-9390-7