
Abstract
We present a case of a 23-year-old man with a tumor containing glial and rhabdoid elements where the former had features of a pleomorphic xanthoastrocytoma (PXA) and the latter had the immunophenotype and genetic profile of an atypical rhabdoid/teratoid tumor. The patient presented with a short history of raised intracranial pressure with rapid deterioration in sensorium. He had a poor outcome despite surgery and radiotherapy. We report this case because of its unusual presentation in adulthood and its occurrence in association with a PXA. We speculate that the PXA was a quiescent tumor and that the secondary genetic alterations, including inactivation of the INI1 gene led to clinical progression.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Malignant rhabdoid tumors are increasingly recognized within the central nervous system (CNS). Initially described in the kidney [1, 2], tumors with similar histology were subsequently identified in many extra-renal sites especially the CNS [3, 4].
Over the past decade, it has become clear that these tumors are highly aggressive with short overall survival. These tumors have deletions and germline or somatic mutations involving the INI1/hSNF5 tumor suppressor gene on chromosome 22q11.2 [5–14]. Central nervous system atypical teratoid/rhabdoid tumor (AT/RT) occurs predominantly in children, while tumors that develop a secondary rhabdoid phenotype (“composite rhabdoid tumors”) are seen more often in adults [15]. Composite rhabdoid tumors generally represent malignant degeneration of other primary tumor types and are usually not associated with INI1/hSNF5 deletions, germline or somatic mutations, or loss of INI1 protein expression. We present an unusual case of a young adult with a tumor containing glial and rhabdoid elements, where the former had features of pleomorphic xanthoastrocytoma (PXA) and the latter had the immunophenotype and genetic profile of AT/RT.
Case report
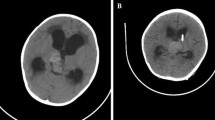
This 23-year-old man presented with altered sensorium for two days. For three weeks prior, he experienced progressively worsening, intermittent headaches with vomiting, double vision, and visual obscuration. He was confused but followed commands. Physical examination revealed bilateral papilledema, a left upper motor neuron facial paresis, and left hemiparesis. There was no neck stiffness and his past medical history was negative for seizures. The magnetic resonance image (MRI) of the brain (Fig. 1a–c) showed a 5 × 6 × 5 cm mass in the right frontal region extending from the cortical surface to the level of the frontal horn of the right lateral ventricle. The latter featured a large area abutting the ventricular surface, but there was no intraventricular extension. Focally the mass included areas that were hyperintense on T1 and hypointense on T2 weighted images, suggestive of recent hemorrhage. There was white matter edema surrounding the lesion and patchy contrast enhancement of the tumor was evident with gadolinium.

Preoperative magnetic resonance images showing a 6 × 6 × 6 cm mass in the left frontal region abutting the frontal horn of the right lateral ventricle. It is hyperintense on the T1 weighted sagittal image, suggestive of a recent hemorrhage (a), with some gadolinium enhancement (b); and it is iso to hypointense on the T2-weighted axial image (c). The images show extensive white matter edema with mass effect
The patient underwent an emergency craniotomy with partial excision of the tumor (Fig. 2a, b). At surgery, the brain was tense and herniated through the dural incision. The tumor was predominantly solid and firm with patchy xanthochromic staining suggestive of prior hemorrhage. Both tumor necrosis and thrombosed vessels were visible and the tumor–brain interface appeared sharp. The right lateral ventricle was entered, but the tumor did not appear to infiltrate along the ependymal lining.

Immediate postoperative computed tomogram scan after the first surgery showing residual tumor
Microscopic sections revealed a tumor with two distinct morphologic appearances, a high grade component consistent with AT/RT and a low grade glioma with features of PXA. The interface between these two elements was fairly sharp (Fig. 3a). The rhabdoid areas were highly cellular and contained patternless sheets of discohesive epithelioid cells that were focally embedded in a dense reticulin network. In foci, the cells were more spindle shaped. Nuclei were oval to reniform with vesicular chromatin and prominent nucleoli (Fig. 3b). The eosinophilic cytoplasm was eccentrically placed and often contained a globular paranuclear inclusion. Several bi- to multi-nucleated rhabdoid forms were present. There were numerous mitoses and occasional foci of necrosis. The PXA component featured pleomorphic astrocytic to mesenchymal-appearing cells with variable nuclear irregularity, nuclear hyperchromasia, and spindled eosinophilic cytoplasmic processes (Fig. 3c). Multinucleated forms were evident and some cells displayed nuclear pseudoinclusions. Occasional cells had clear vacuoles (xanthomatous features). Mitotic figures were hard to find and there was no endothelial proliferation or necrosis. Scattered eosinophilic granular bodies were seen (Fig. 3d). There were no Rosenthal fibers or dysmorphic neurons. Several foci of lymphocytic vascular cuffing were noted. Reticulin deposition was mildly increased.

Photomicrograph of H & E-stained sections of the tumor showing the sharp interface between the pleomorphic xanthoastrocytoma (PXA) and the AT/RT elements (a); Discohesive sheets of rhabdoid cells in the AT/RT element with eccentrically placed oval to reniform vesicular nuclei and prominent nucleoli (b); The pleomorphic astrocytic forms in the PXA (c); Scattered eosinophilic granular bodies (d).
Immunohistochemical stains showed strong vimentin and smooth muscle actin (SMA) expression in the AT/RT component. Focally there was also membranous immunoreactivity for epithelial membrane antigen (EMA) and CD34. Staining for INI1 protein showed loss of nuclear expression in the majority of AT/RT tumor cells (Fig. 4a). In contrast, there was retained nuclear expression in the PXA component (Fig. 4b) and within intratumoral endothelial cells of both elements. Scattered nuclei were positive for p53 protein. The Ki-67 stain depicted a markedly elevated proliferation index in the AT/RT region, but a low level in the PXA portion (Fig. 4c). Strong GFAP (Fig. 4d) and vimentin positivity was seen in the latter, whereas most AT/RT cells were negative (Fig. 4e). There were clusters of CD34 positive cells with highly ramified cell processes in the PXA component and neurofilament protein (NFP) highlighted entrapped axons, indicating that the white matter was infiltrated. NFP expression was also seen in rare rhabdoid cells. The PXA component was negative for SMA.

Immunohistochemical labeling showing the AT/RT element with loss of nuclear expression of INI1 (a) and the PXA element with retained nuclear expression of INI1 (b); Contrasting Ki-67 labeling indices in the two elements (c), with high Ki-67 labeling index in the AT/RT (lower half) and low Ki-67 labeling index in the PXA (upper half); Expression for GFAP in the PXA (d) and negativity for GFAP in most of the rhabdoid cells (e)
Fluorescence in situ hybridization (FISH) was performed to investigate the status of the INI1 gene on chromosome 22q as previously reported [15]. Probes directed against the BCR (22q11.2; nearby to the INI-1 locus), and the NF2 (22q12) genes were employed. Both rhabdoid and glial areas were analyzed. FISH revealed one signal for both markers in most rhabdoid tumor cells, suggesting either a large 22q deletion or a monosomy 22 (Fig. 5a). In contrast, approximately 50% of the cells from the glial component had two copies of chromosome 22, whereas the remaining cells had varying numbers (3–8) of the chromosome 22 probes, consistent with gains of chromosome 22 (Fig. 5b).

FISH images of the AT/RT element showing 22q deletion with one green and one red signal (a) and the PXA element showing polysomy 22q with > 2 green and > 2 red signals (b)
Mutation analysis of the INI1 gene was performed as previously reported [14] using DNA isolated separately from the rhabdoid and the PXA components of this patient’s formalin fixed paraffin embedded tumor. A cytosine to thymidine substitution was detected at position 472 in codon 158 in exon 4 of the INI1 gene in the rhabdoid tumor area. However, the mutation was not detected in the PXA component. This mutation is predicted to result in change from an arginine to a stop codon, resulting in a prematurely truncated protein.
Based on the initial biopsy report of a high grade astrocytoma with rhabdoid features, the patient was started on radiation therapy (RT) to the head. However a month into the course of RT he developed a progressive left sided weakness and became drowsy. The CT scan showed that the tumor had increased in size, with marked cerebral edema and mass effect (Fig. 6a, b); therefore he underwent a repeat craniotomy and a radical excision of the tumor. Radiation therapy was resumed during which he had a fluctuating sensorium with hyponatremia and chest infection. His CT scan showed no residual tumor. He was discharged at request and died two weeks later at home, probably due to the chest infection.

Computed tomogram scans during radiation therapy showing an increase in the edema and size of the tumor
Discussion
The rhabdoid phenotype is defined by large tumor cells with eccentrically placed vesicular nuclei containing prominent nucleoli, eosinophilic cytoplasm, and paranuclear inclusions. The term rhabdoid reflects the fact that despite their resemblance of rhabdomyoblasts, these cells typically lack evidence of true skeletal muscle differentiation. [1, 16]
There are essentially two groups of rhabdoid tumors. The truly distinctive tumor entity called malignant rhabdoid tumor (MRT) outside the CNS or atypical teratoid/rhabdoid tumor (AT/RT) within the CNS is seen in the pediatric population, has a polyphenotypic immunoprofile and characteristic deletions and mutations (somatic or germline) involving the INI1/hSNF5 tumor suppressor gene on chromosome 22q11.2; it has a highly aggressive biology with short patient survival times [5–14]. The other category, the composite rhabdoid tumor is seen most often in adults as a secondary phenotype within a parent neoplasm. These tumors do not share the genetic alterations of the first group and bear clear phenotypic and genotypic allegiance to the parent neoplasm. As such, they do not show evidence of INI1 gene inactivation [15, 17–23].
The main differential diagnosis in the present case was between that of a ‘composite rhabdoid tumor’ (CRT) arising in a high grade astrocytoma (e.g., glioblastoma) versus a mixed tumor containing a true AT/RT component. The latter was found to be the case and the adjacent glial element showed features of PXA. The diagnosis of AT/RT was supported by a polyphenotypic immunoprofile, loss of INI1 expression, evidence of a 22q deletion by FISH, and a detectable INI1 gene mutation. In contrast, none of these were found in the PXA region.
The case is highly unusual on several counts. First, the presentation in adulthood (albeit early adulthood) is uncommon in AT/RT. Second, PXA is a rare tumor itself and to our knowledge has never been reported in combination with AT/RT. Although the interface between these two elements was fairly discrete, there was a suggestion of a transition zone between the rhabdoid and glial areas focally, with a slight intermingling of the two cell types, suggesting that the AT/RT component might have nevertheless arisen from the PXA. If so, given that the AT/RT cells had monosomy 22q, the rhabdoid tumor would most likely have arisen from the diploid cells in the PXA, rather than the cells with multiple copies of chromosome 22. By analogy, Allen et al. [24] recently reported a ganglioglioma that developed an AT/RT component after 10 years; the histologic and genetic data was fairly similar to that of the current case. Consequently, it seems less likely that the two components of the present tumor arose from divergent differentiation of a primitive progenitor cell type. Furthermore, it is possible that the PXA was the initial quiescent tumor and that secondary genetic alterations, including the inactivation of the INI1 gene led to clinical progression. Although both low and high grade areas were present, the high grade element naturally dictated the ultimate prognosis for this patient.
In summary, we have presented a case of an AT/RT that likely arose from a low grade precursor neoplasm, a PXA. Recognition of this rare complication is important, as these patients subsequently progress rapidly and may benefit from more aggressive forms of therapy.
References
Weeks DA, Beckwith JB, Mierau GW et al (1989) Rhabdoid tumor of kidney. A report of 111 cases from the National Wilms’ Tumor Study Pathology Center. Am J Surg Pathol 13:439–458
Beckwith JB (1983) Wilms’ tumor and other renal tumors of childhood: a selective review from the National Wilms’ Tumor Study Pathology Center. Hum Pathol 14:481–492
Rorke LB, Packer RJ, Biegel JA (1996) Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg 85:56–65
Rorke LB, Packer R, Biegel J (1995) Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood. J Neurooncol 24:21–28
White FV, Dehner LP, Belchis DA et al (1999) Congenital disseminated malignant rhabdoid tumor: a distinct clinicopathologic entity demonstrating abnormalities of chromosome 22q11. Am J Surg Pathol 23:249–256
Versteege I, Sevenet N, Lange J et al (1998) Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 394:203–206
Rousseau-Merck MF, Versteege I, Legrand I et al (1999) hSNF5/INI1 inactivation is mainly associated with homozygous deletions and mitotic recombinations in rhabdoid tumors. Cancer Res 59:3152–3156
Sevenet N, Sheridan E, Amram D et al (1999) Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am J Hum Genet 65:1342–1348
Judkins AR, Mauger J, Ht A et al (2004) Immunohistochemical analysis of hSNF5/INI1 in pediatric CNS neoplasms. Am J Surg Pathol 28:644–650
Hoot AC, Russo P, Judkins AR et al (2004) Immunohistochemical analysis of hSNF5/INI1 distinguishes renal and extra-renal malignant rhabdoid tumors from other pediatric soft tissue tumors. Am J Surg Pathol 28:1485–1491
Biegel JA, Fogelgren B, Wainwright LM et al (2000) Germline INI1 mutation in a patient with a central nervous system atypical teratoid tumor and renal rhabdoid tumor. Genes Chromosomes Cancer 28:31–37
Biegel JA, Kalpana G, Knudsen ES et al (2002) The role of INI1 and the SWI/SNF complex in the development of rhabdoid tumors: meeting summary from the workshop on childhood atypical teratoid/rhabdoid tumors. Cancer Res 62:323–328
Biegel JA, Tan L, Zhang F et al (2002) Alterations of the hSNF5/INI1 gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin Cancer Res 8:3461–3467
Biegel JA, Zhou JY, Rorke LB et al (1999) Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res 59:74–79
Perry A, Fuller CE, Judkins AR et al (2005) INI1 expression is retained in composite rhabdoid tumors, including rhabdoid meningiomas. Mod Pathol 18:951–958
Beckwith JB, Palmer NF (1978) Histopathology and prognosis of Wilms tumors: results from the First National Wilms’ Tumor Study. Cancer 41:1937–1948
Shimazaki H, Aida S, Sato M et al (2001) Lung carcinoma with rhabdoid cells: a clinicopathological study and survival analysis of 14 cases. Histopathology 38:425–434
Perry A, Scheithauer BW, Stafford SL et al (1998) “Rhabdoid” meningioma: an aggressive variant. Am J Surg Pathol 22:1482–1490
Oshiro Y, Shiratsuchi H, Oda Y et al (2000) Rhabdoid features in leiomyosarcoma of soft tissue: with special reference to aggressive behavior. Mod Pathol 13:1211–1218
Levine PH, Mittal K (2002) Rhabdoid epithelioid leiomyosarcoma of the uterine corpus: a case report and literature review. Int J Surg Pathol 10:231–236
Knapik J, Yachnis AT, Ripley D et al (2001) Aggressive uterine sarcoma with rhabdoid features: diagnosis by peritoneal fluid cytology and absence of INI1 gene mutation. Hum Pathol 32:884–886
Kepes JJ, Moral LA, Wilkinson SB et al (1998) Rhabdoid transformation of tumor cells in meningiomas: a histologic indication of increased proliferative activity: report of four cases. Am J Surg Pathol 22:231–238
Gokden N, Nappi O, Swanson PE et al (2000) Renal cell carcinoma with rhabdoid features. Am J Surg Pathol 24:1329–1338
Allen JC, Judkins AR, Rosenblum MK et al (2006) Atypical teratoid/rhabdoid tumor evolving from an optic pathway ganglioglioma: case study. Neuro–oncol 8:79–82
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chacko, G., Chacko, A.G., Dunham, C.P. et al. Atypical teratoid/rhabdoid tumor arising in the setting of a pleomorphic xanthoastrocytoma. J Neurooncol 84, 217–222 (2007). https://doi.org/10.1007/s11060-007-9361-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-007-9361-z