
Abstract
New niobium-based oxygen carriers were prepared adopting polyol-mediated process. Four different polyols were used (diethylene glycol, triethylene glycol, tetraethylene glycol, and 1,2-propanediol) and four different quantities of water (0 mL, 1 mL, 10 mL, 30 mL, with a total volume of 80 mL of solution). Series of bulk and alumina-supported materials were prepared. Different structural and textural properties were obtained, depending on polyol type and hydrolysis ratio. After thermal treatment at 1000 °C, alumina-supported materials were evaluated in a lab-scaled fixed bed reactor. Despite a low conversion of methane due to a low content of active phase AlNbO4, they presented a high selectivity to syngas (a mixture of H2 and CO) during the first few seconds of reaction with CH4, indicating a high selectivity towards chemical-looping reforming process.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Chemical looping is an excellent alternative technology for industrial processes that make use of fossil fuel, (Kvamsdal et al. 2007; Miracca et al. 2009; Adanez et al. 2012) such as combustion reactions that generate energy and reforming reactions that generate hydrogen. One of the best advantages of this technology is that it can capture the CO2 that is inherently produced by these processes. Chemical-looping combustion (CLC) and chemical-looping reforming (CLR) use an oxygen carrier, a metal oxide which may be supported on an inert support matrix (alumina, for example) (Gayán et al. 2008; de Diego et al. 2005; Zafar et al. 2006) and have characteristics similar to that of an heterogeneous catalyst.
This oxygen carrier material circulates through two reactors: an air reactor and a fuel reactor, as seen in Fig. 1 (Adanez et al. 2012). When in contact with the fuel, the metal oxide provides oxygen. The reaction is either total oxidation (combustion) or partial oxidation (reforming). When the material enters the air reactor, it is either partially or totally reduced. There, it is reoxidized, leading to a continuous process of combustion or reforming of the fossil fuel.

Diagram of the interconnected reactors
Chemical-looping technology is still under development. There are many technological challenges ahead before it can be implemented, such as the need to scale up the reactors that are currently used in oxygen carrier studies. In the specific case of the CLR process, one of the technological challenges that limits its development is finding an oxygen carrier that is highly selective to the reforming reaction at high temperatures (the process normally takes place between 800 and 1000 °C).
Niobium oxide
Over the past few years, the scientific community has begun to consider niobium oxide as a good alternative in heterogeneous catalysis (Tanabe 2003; Ziolek 2003) because it is acidic and has redox and photocatalytic properties. Nb has different oxidative states from − 1 to +5. It can form NbO, Nb2O3, NbO2, and Nb2O5 oxides. Nb2O5 is the most common, the Nb+5 oxidation state being the most stable (Nowak and Ziolek 1999).
Different thermal treatments can cause Nb2O5 to develop different crystalline structures. The most common phases are amorphous Nb2O5, TT-Nb2O5 (pseudo-hexagonal structure), T-Nb2O5 (orthorhombic structure), and H-Nb2O5 (monoclinic structure) (Ko and Weissman 1990). Many studies have been carried out regarding the application of this oxide in heterogeneous catalysis, with the aim of taking advantage of this crystallographic versatility. Niobium oxide can be used as a support (Li et al. 2004; Mozer and Passos 2011), promoter (Chun et al. 2010), or even as active phase (Li et al. 2005; He et al. 2013) in a catalyst. It can even influence the properties of known catalysts, increasing their life cycle and/or reactivity (Tanabe 2003).
The polyol process
Another scientific area that has recently attracted increasing attention is the study of nanostructured materials. These materials are promising in the area of heterogeneous catalysis because they have not only higher surface areas, but also different properties than bulk materials due to quantum size effects that allow controlling their band gap.
Several groups have explored different methods for the synthesis of nanostructured niobium oxide, producing materials with different particle sizes and morphologies (Zhao et al. 2012). In this study, we have adopted the polyol method, in which a salt precursor of the oxide is solubilized in a α-diol or a glycol ether to which a small amount of water is added. Under these conditions, the oxide is synthesized through forced hydrolysis, similar to the sol-gel process. The solvent has a large number of hydroxyl groups that strongly interact with the surface of the particle during its formation, and in the end, a nanostructured particle with controllable shape and dimension is obtained (Fiévet and Brayner 2013).
The objective of this study is to synthesize and characterize niobium oxides prepared from a polyol process (with or without an alumina support) and to evaluate the applicability of the supported and calcined materials in the CLR process.
Experimental
Preparation
Commercial Nb2O5 (HY-340) and the precursor ammonium niobate oxalate were supplied by CBMM (Companhia Brasileira de Metalurgia e Mineração, Brazil); the alumina (Puralox NWa 155) was kindly provided by Sasol (Germany).
Synthesis was carried out in two different ways. Niobium oxide was prepared with and without an alumina support. For synthesis without the support, the oxides were prepared by the hydrolysis of 1 g of ammonium niobiate oxalate in a polyol medium using a small quantity of zinc acetate (0.0015 g) as nucleant. Four different polyols were used: diethylene glycol (DEG), triethylene glycol (TEG), tetraethylene glycol (TTG), and 1,2-propanediol (1,2PD). Different amounts of water were added to induce the hydrolysis process, always maintaining a total volume of 80 mL. For the preparation of the supported materials, ammonium niobiate oxalate and alumina were added to the polyol/water mixture, so as to produce 5 wt% Nb2O5/Al2O3 samples. The water/polyol ratios were identical to those used for the synthesis of the unsupported samples. Reference code of each sample is related to their synthesis parameters. Table 1 shows the relation between reference code and synthesis parameters.
The suspensions were maintained for both series of materials under stirring. The mixtures were then heated to the boiling point of the polyol-water mixture. This temperature varies over a wide range of values depending mostly on the volume of water in the solution and also on the polyol type (see Table 1 for temperature values). These conditions were maintained for 3 h with reflux. The resulting materials were centrifuged, washed with ethanol, and dried at 50 °C. Supported materials were calcined, in order to stabilize this material for evaluation tests, to 1000 °C for 3 h, using a heating rate of 3 °C/min, under air in a furnace. When calcined, samples receive a “C” at the end of their reference (for example, sample prepared with diethylene glycol, with 10 mL of water and supported (DEG 10 Al), when calcined, is called DEG 10 Al C). Table 1 shows sample names and their synthesis conditions adopted.
Characterizations
Materials were characterized by X-ray diffraction (PANalytical X’PertPRO, with CoKα radiation, in the range of 10° < 2θ < 80°), transmission electron microscopy (TEM) observations, and EDS X-ray microanalysis were carried out on a JEOL JEM 2100Plus transmission electron microscope, operating at 200 kV, interfaced to Oxford Instruments AZtec EDS system with an X-Max T large area (80 mm2) SDD detector, UV-visible diffuse reflectance (Perkin Elmer Lambda 1050), nitrogen adsorption (surface area calculated by the BET method, Quantachrome Nova 2200), and thermogravimetry with differential thermal analysis (SETARAM). TG-DTA analyses were carried out in an air atmosphere from room temperature to 950 °C at a heating hate of 10 °C/min. Acidity was evaluated by FTIR of adsorbed pyridine. FTIR spectra were obtained on a Bruker Vector 22 FT-IR spectrometer (resolution, 4 cm−1). Samples were pressed into self-supported wafers (ca. 10 mg/cm2) and heated at 400 °C for 2 h in O2 (PO2 = 13.3 kPa). They were evacuated afterward for 1 h at this temperature. Pyridine (P = 133 Pa) was introduced at RT and spectra were recorded after thermal desorption either at 150 or at 350 °C. Only Lewis acid sites were detected. The number of Lewis acid sites remaining after thermal treatment at 150 °C (corresponding to weak and strong Lewis sites) and after thermal treatment at 300 °C (corresponding to strong Lewis sites) was calculated by measuring the area of the band at 1454 cm−1 after thermal treatment at these two temperatures and using the following equation (Barzetti et al. 1996; Kitano et al. 2012):
in which qH is the concentration of acid sites in the sample; AI is the area under the peaks in the IR analysis; R is the radius of the disk (0.8 cm); w is the mass of the sample; and εI is the absorption coefficient (2.22 cm/μmol for the band at 1454 cm−1) (Emeis 1993).
In-situ XRD thermal analyses in an air atmosphere were conducted on non-supported materials prepared with 10 mL H2O and supported sample TTG 10 Al. The experiments were carried out from room temperature to 950 °C. The XRD profiles were obtained in all temperatures with 100° steps.
Evaluation
Supported oxygen carriers calcined at 1000 °C were evaluated in the CLR process. In a CLR process, two reactors are used: a fuel reactor and an air reactor. To simulate this set-up, reduction cycles with methane and reoxidation cycles with oxygen were carried out in a single reactor (in quartz) at 950 °C. One gram of supported oxygen carrier was used with a total gas flow of 50 mL/min, under atmospheric pressure. The gas contained 10% CH4 for the reduction step and 10% O2 for the oxidation step. The remainder of the gas was argon in both cases. Reaction times were set at 6 min for the reduction step and 12 min for the oxidation step.
The reaction products were analyzed with an MKS e-Vision+ mass spectrometer and by a Varian CP-3800 gas chromatograph. The results were evaluated considering the selectivity of the CLR and CLC processes, carbon deposit on the surface of the material, and methane conversion throughout the reaction. Reactions represented in equations from 2 to 4 were considered to calculate selectivities and conversion.
From these reactions, several hypotheses should be made:
-
(a)
According to Eq. 2, all CO formed can be related to CLR reaction (1 mol of CO/mol of CH4);
-
(b)
According to Eq. 3, all CO2 formed can be related to CLC reaction (1 mol of CO2/mol of CH4);
-
(c)
According to Eq. 4, carbon deposition can be estimated by subtracting to total H2 formed, the fraction of H2 formed according to Eq. 2 (2 mol H2/mol CO). Equation 5 can be written:
where [H2] is the total hydrogen produced and [H2(CLR)] is the hydrogen produced from Eq. 2. [H2(CLR)] can be estimated as:
where [CO] is the total amount of CO produced. Then the contribution from carbon deposition could be estimated as:
CLC and CLR contributions could be estimated as:
Then, selectivities can be calculated as:
Conversion of methane could be calculated as:
where [CH4(not reacted)] is the amount of CH4 not reacted, directly identified from evaluation curves.
Results and discussion
Non-supported samples
Table 2 shows the main characterization results obtained for these samples.
Variation in the volume fraction of water has some influence on the structure of the material, as seen in Fig. 2, which shows the X-ray diffraction profiles of uncalcined samples prepared using various water/tetraethylene glycol (TTG series) ratios (see Table 1 for details). Materials prepared with 10 and 30 mL of water presented HNbO3 phase (ICDD card 00-036-0794). On the other hand, materials prepared with 1 mL of water appears to have an amorphous structure, similar to that observed for commercial oxide HY 340. This could indicate that an increase in water volume can lead to materials with more organized structures. The diffraction profiles of materials prepared with 10 mL and 30 mL of water show that with even larger amounts of water, the material begins to have a slightly less ordered structure (the peaks on the material prepared with 10 mL of water are slightly better defined than those of the material prepared with 30 mL of water).

X-ray diffraction profiles of unsupported samples before calcination: TTG 01 (red), TTG 10 (green), and TTG 30 (blue). The profile of a commercial Nb2O5 sample (HY 340, black) is also shown, for comparison. The lines identified as HNbO3 phase (ICDD card 00-036-0794) are marked with an asterisk
TG-DTA together with in situ XRD profiles recorded during thermal treatment reveal the transformations that take place in the materials as the temperature increases.
TG curves of a series of non-supported material prepared with 10 ml of water in the reaction mixture and using various polyols are reported in Fig. 3a. On all curves, there are two main regions of mass loss: the first is from room temperature to ca. 170 °C and the second is between ca. 170 and 300 °C. The DTA curves (Fig. 3b) show that the first mass drop corresponds to an endothermic event (and is therefore likely due to the loss of hydration water). The second drop occurs at slightly different temperatures depending on the polyol and has different heat releases (in the case of samples TEG 10 and TEG 30, not shown, there are no large variations in energy associated with this second mass loss). A third mass loss can be seen for most samples between 550 and 620 °C. It is weaker than the second mass loss and associated with an exothermic peak.

Thermogravimetric analysis of uncalcined non-supported samples: DEG 10 (black), TEG 10 (red), TTG 10 (blue), and 1,2PD 10 (green). a TG profiles. b DTA profiles
The in situ XRD profiles recorded during calcination reveal modifications to the crystalline structure at temperatures of 300 °C, 600 °C, 800 °C, and 950 °C, as seen in Fig. 4.

In situ XRD analysis of TEG 10. a Diffraction profiles (between 2θ = 20 and 2θ = 40°) at all temperatures analyzed and b full diffraction profiles at structural modification temperatures (corresponding to profiles in red on (a) (the peaks marked with an asterisk correspond to the corundum phase and are due to the corundum wafer on which the sample is deposited for the in situ XRD measurement)
At a temperature of 300 °C, the first change in the structure of the materials takes place, with the loss of the diffraction lines of the HNbO3 phase. The alteration coincides with the loss of mass seen in the TG curves between 170 and 300 °C. This mass loss can be only partially explained by the decomposition of HNbO3 into amorphous niobium oxide (Nb2O5.nH2O). Indeed, decomposition of HNbO3 in Nb2O5.nH2O can represent a maximum mass loss of approximately 6% (considering n = 0). However, as it can be seen, the mass loss for all samples is higher than 20%. Another event that could be happening at the same time is the thermal decomposition of the small part of polyol that had not been eliminated during washing step. The decomposition of polyols could explain why this second weight loss do not occur at the exact same temperature for samples prepared with different polyols. A similar weight loss has previously been reported by Li et al. (2016) during thermal treatment of niobium oxohydroxide prepared in ethanol and assigned to the decomposition of adsorbed organic molecules. The identification of the niobium oxide phase present after thermal treatment at 300 °C is not possible as the peaks are very broad and lack intensity. This is thought to be an indication that an amorphous structure of the oxide Nb2O5 forms after 300 °C. Another event that takes place at this temperature is the appearance of sharp peaks, which are due to the corundum wafer on which the sample had been deposited for measurement. Because of the mass loss, a contraction of the sample has occurred, leading the exposure of the corundum wafer to the X-ray beam.
The second modification takes place at 600 °C, where Nb2O5 with a pseudo-hexagonal structure, is observed (ICDD card 00-028-0317). This transformation coincides with the thermal event in the DTA curves between 550 and 620 °C discussed above. The third changed is observed after thermal treatment at 800 °C, where the structure changes from pseudo-hexagonal to orthorhombic (ICDD card 00-30-0873). Figure 5 shows an enlargement (in the 30-45° 2θ range) of the diffraction profiles recorded after thermal treatment at 600 and 800 °C: the phase transformation is evidenced by the splitting of the diffraction lines on the profile recorded after thermal treatment at 800 °C.

Diffraction profiles in the 30–45° 2θ range recorded at 600 °C (with diffraction lines characteristic of the pseudo-hexagonal structure) and at 800 °C (with diffraction lines characteristic of the orthorhombic structure). Sample: TEG 10
These results agree with data from Ko et al. who investigated the thermal transformations of Nb2O5 (Ko and Weissman 1990). According to the work of these authors, the low temperature phase (pseudo-hexagonal structure, called TT-Nb2(O,Y)5 + n) and intermediate temperature phase (orthorhombic structure, called T-Nb2O5) have similar crystallographic structures but differ by the fact that the TT-Nb2(O,Y)5 + n phase contain impurities (OH−, for example). Due to these impurities in the TT-Nb2(O,Y)5 + n, the Nb atoms can indifferently occupy two crystallographically similar sites, whereas, in the orthorhombic structure, they occupy either one or the other. This explains why the peaks for the pseudo-hexagonal structure are broader (and also the splitting of the peaks in the orthorhombic structure) (Ko and Weissman 1990).
The last change takes place at a temperature of 950 °C, when the monoclinic phase forms. This last transformation does not take place for sample TTG 10, which maintains the orthorhombic structure until 950 °C.
An interesting point is that we could not detect any signal in the DTA curves that would indicate modifications in the crystalline structure around 800 °C (transition from TT-Nb2O5 to T- Nb2O5) and around 950 °C (transition from T- Nb2O5 to H- Nb2O5).
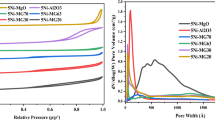
Figure 6 a shows the high-resolution transmission electron microscopy (HRTEM) micrograph of the TEG 10 sample. We can see that the sample is amorphous (the SAED pattern in insert) which confirms the XRD result at 25 °C. Figure 6 b shows the elemental analysis (EDX) of the sample proving the presence of niobium and oxygen. The structural properties of the samples were also investigated by nitrogen physisorption. BET surface areas, pore sizes, and pore volumes for the non-supported samples are compiled in Table 2. The specific areas vary over a wide range of values, depending on the water volume and on the polyol, whereas the pore size distribution is almost identical when the same polyol is used. The N2 adsorption-desorption isotherms of a series of samples prepared with various polyols and the same amount of added water (30 mL) are reported in Fig. 7. The samples prepared with triethyleneglycol (TEG) and tetraethyleneglycol (TTG) have type II isotherms according to IUPAC classification (IUPAC 1985, 1994), indicating that the materials prepared with these two polyols are not porous. The high surface area, which reached 332 m2/g for sample TTG 01, is related to the nano-scale size of the particle formed.

a TEM image and SAED pattern in insert. b EDX spectrum corresponding to TEG 10 sample

N2 adsorption–desorption profiles of non-supported and non-calcined materials prepared with 30 mL of water added to various polyols. (▲) DEG 30, (●) TEG 30, (♦) TTG 30, (▼)1,2PD 30. The isotherm of commercial oxide (■) HY 340 is also shown for comparison
Materials prepared with 1,2-propanediol, as well as commercial oxide HY 340, present a combination of type I and type IV isotherms with a H2 type hysteresis loop, according to IUPAC classification (IUPAC 1985, 1994). These characteristics are indicative of a microporous and mesoporous structure with ink bottle-shaped isotherm. The average mesopores size, calculated by applying the BJH model to the adsorption isotherm is 3.3 nm.
The three samples prepared with diethylene glycol behaved differently, depending on the water content. Samples DEG 01 and DEG 30 had types I + II isotherms. Sample DEG 01 had a characteristic type H1 hysteresis characteristic of cylindrical pores, while sample DEG 30 had type H2 hysteresis, indicating that the pores had an “Ink Bottle” shape. Sample DEG 10 has a very small surface area, indicating the absence of meso- or micropores in this sample.
The UV-Vis spectra of a series of samples prepared without alumina support and using a solution of TTG added with various amounts of water are shown on Fig. 8. No visible trend in the evolution of the spectra with water content can be observed. Unlike commercial niobium HY 340, all of the synthesized materials have a signal in the visible zone (between 400 and 700 nm). This signal may be caused by oxygen vacancies in the structure of the samples, giving them a caramel color, as reported by Brayner and Bozon-Verduraz (2003).

UV-Vis spectra of a series of uncalcined non-supported materials prepared using tetraethylene glycol
Supported samples
Synthesis parameters for alumina-supported niobium oxide samples are reported in Table 1. These samples were characterized using XRD and HRTEM analyses to determine their morphologies. Table 3 shows the main characterization results obtained for these samples.
XRD profiles for the supported materials before calcination of all samples are the same as the one of NWa 155 (γ-Al2O3 phase). The phase of niobium formed could not be identified, either simply because the level was below the detection limit of the technique (the samples contain ca. 5 wt% of Nb2O5), or even because the high dispersion of the niobium oxide phase on the γ-alumina support result in the formation of a monolayer. In both cases, niobium phase could not be detected by means of an X-ray diffractometry.
The modification of the crystallinity of TTG 10 Al sample during calcination was investigated in situ using XRD (Fig. 9). Crystallization of Nb2O5 (pseudo-hexagonal phase, TT-Nb2O5) is observed 700 °C with the presence of two small peaks at 26.4° and 33.3° (corresponding to the peaks with relative intensity of 90 and 100%, respectively according to ICDD card 00-028-0317). This structure might appear because of sintering of the phase that contains niobium. Then, larger oxide particles are formed, which makes them detectable. After 900 °C, the two weak peaks associated with the Nb2O5 phase disappear, and new diffraction peaks are observed—29.4° and 33.7°—that can be assigned to the aluminum niobate (AlNbO4) phase (ICDD card 00-26-030), showing the strong interaction of the active phase with the support. It shows that the active phase for evaluation tests is not the Nb2O5, but the mixed oxide formed from this strong interaction. Moreover, at this temperature, a transition of the alumina phase from gamma to delta is observed.

a In situ XRD analysis of TTG 10 Al. Profiles in red: Temperatures where the crystalline structure has been modified. b Zoom between 20° and 40° (2θ)
In comparison with the TEG 10 sample (Fig. 6), the TEG 10 Al sample was analyzed by TEM (Fig. 10a, b) and EDX (Fig. 10c, d). The morphology of alumina is observed (Fig. 10 a, b). The EDX analysis proves the presence of niobium in both cases. The peak is small because we only have 5 wt% in niobium in the sample.

a, b TEM images. c, d EDX spectra respectively for TEG 10 Al sample
To better characterize the TEG 10 Al sample, STEM-HAADF and STEM-EDX map analyses were performed (Fig. 11). Figure 11 a shows the area that was analyzed. Figure 11b shows the STEM-HAADF result of the area that was chosen. Figure 11c–e show the mapping of the zone (11c (Nb-L), 11d (O-K), and 11e (Al-K)). It can be seen that niobium is well dispersed throughout the alumina (Fig. 11c) and finally Fig. 11f presents the EDX analysis result of the TEG 10 Al sample which shows the presence of the niobium peak.

a TEM and b STEM-HAADF images. STEM-EDX c Nb-L, d O-K, e Al-K maps. f EDX sum spectrum of the map for TEG 10 Al sample
Nitrogen physisorption results for the uncalcined samples are shown on Table 3. The surface area is practically the same as that of the alumina support (NWa 155). It is to be expected considering the low niobium loading in all samples.
Energy gap values of uncalcined samples are reported in Table 2 (samples prepared without alumina) and Table 3 (samples prepared with alumina). They were estimated using diffuse reflectance UV-Vis spectroscopy. The energy gap values for the synthesized samples are very close to those of commercial oxide HY 340 (3.3 eV). The energy gap values do not follow a clear trend and fluctuate from 3.2 to 3.7 eV depending on the sample. For sample TTG 00 Al, that did contain niobium, no energy gap value could be measured. A possible explanation for this is the presence of small amounts of polyol adsorbed on the surface of the materials.
The acidic properties of one of the supported samples (TTG 10 Al) were investigated by means of pyridine desorption detected by FTIR. Only Lewis acid sites were detected (band at 1454 cm−1). The number of Lewis acid sites remaining after thermal treatment at 150 °C (corresponding to weak and strong Lewis sites) and after thermal treatment at 300 °C (corresponding to strong Lewis sites) are reported in Table 4 for TTG 10 Al and the alumina support alone (NWa 155 sample). Alumina alone and TTG 10 Al have similar acidities. The acidic properties of TTG 10 Al are therefore due mostly to its alumina component, and the small fraction of niobium added in this sample does not induce significant modification in the acidity.
Characterization results of supported niobium oxide presented in Table 3 showed that no significant differences were observed for different oxygen carriers. Differences have been observed only for non-supported oxides. Thus, only one of the oxygen carriers was selected to evaluations tests. To select this carrier, we considered the polyol cost and final temperature for reaction, assuming that costs for synthesis should be as low as possible. By these criterions, a good alternative is carrier DEG 10 Al C, prepared with diethylene glycol, the cheapest polyol among those studied here. Moreover, because of its high-water content, temperature during synthesis process was only 134 °C.
Fixed bed reactor evaluation
Figure 12 shows the results of reduction with 10% CH4/Ar and oxidation with 10% O2/Ar for DEG 10 Al C (DEG 10 Al calcined at 100 °C) sample. The result refers to the fifth redox cycle carried out with this material. For the reduction step (the first 6 min), as soon as the methane is sent, a large amount of H2 is formed; together with CO and a small amount of water, unreacted methane is also present in the effluent. CO2 is not produced during the reduction reaction.

Fifth redox cycle for carrier DEG 10 Al C
After this reduction step, the reactor is purged under Ar during 5 min in order to remove all gases, and the oxidation step is started by flowing the O2/Ar mixture on the sample. A large amount of CO is formed during the oxidation step. This shows that during the reduction step, a large amount of methane was converted into carbon deposit on the surface of the material and hydrogen was released. This reaction can be confirmed by the high H2/CO ratio after the ten first seconds of reaction.
Selectivity curves for the Chemical-Looping combustion (CLC) and Chemical-Looping Reforming (CLR) reactions and carbon deposit processes were constructed (see experimental part for details). These reactions are respectively represented by Eqs. 14, 15, and 16.
Figure 13 shows the selectivity and methane conversion results throughout the reduction step. During the first 10 s of reaction, the CLR process is completely selective although methane conversion is very low, reaching a maximum of 20% of the methane sent. After 10 s, the carbon deposit reaction increases gradually in prevalence and becomes the main reaction after 1 min time on stream.

Selectivity and conversion comparison for DEG 10 Al C during reduction step
Compared with other common oxygen carriers applied to chemical-looping reforming, this Nb-based carrier presented a different behavior. Zafar et al. (2005) studied Cu-, Mn-, Ni-, and Fe- oxides supported on SiO2 as oxygen carriers. All of them start the reduction step by the production of CO2 and steam; CO and H2 are produced only after ca. 40 s under reductive conditions, when the oxidation of CH4 to CO2 is no longer total. Ni-based oxygen carrier showed the best reactivity.
Pröll et al. (2010) investigated the reactivity of Ni-based oxygen carrier on a 140-kW pilot plant. Results show that a mixture of CO2, H2O, CO, and H2 is expected from the fuel reactor stream in different conditions of air to fuel ratio.
Figure 14 shows the selectivity and conversion profile from a Ni-based oxygen carrier containing 19% of NiAl2O4 under the same conditions as described for Fig. 13. Preparation and characterization of such material were previously presented in (Barbosa et al. 2016). Conversion is almost total during all 6 min of reduction step. However, selectivity to CO2 and H2O formation is high at the beginning of reduction, with an increasing in syngas production selectivity a few seconds later. These results are in accordance with Zafar et al. (2005) and Pröll et al. (2010) as discussed here.

Selectivity and conversion comparison for NiAl2O4/Al2O4 oxygen carrier during reduction step
In the present work, we show that Nb-based oxygen carriers presenting AlNbO4 as active phase can generate syngas from the beginning of reduction step without CO2 formation. With an increasing of its reactivity, it could lead to an ideal operational condition where methane is totally converted to generate syngas, increasing the process yield.
Conclusions
Niobium oxide particles were prepared using the polyol method; most of the syntheses resulted in materials containing HNbO3. The crystalline structures vary widely with the water concentration in the system. The materials prepared with 10 mL of water had the clearest peaks on the X-ray diffraction profiles.
For the materials prepared in the presence of the alumina support Puralox NWa 155, AlNbO4 was detected before calcination, indicating a strong interaction between niobia and alumina at high temperatures. When the reaction conditions of the CLR process are simulated, the process is totally selective toward CLR for the initial seconds of the reaction (no CLC products), a result that has never been published before. However, little methane is converted in this region, reaching a maximum of only 20%. After ca. 10 s time on stream, the conversion toward CLR progressively decreases due to the formation of coke. These are only preliminary results that show a great possibility of application for Nb-based oxygen carrier for CLR process.
References
Adanez J, Abad A, Garcia-Labiano F, Gayan P, de Diego L (2012) Progress in chemical-looping combustion and reforming technologies. Prog Energy Combust Sci 38:215–282
Barbosa RD, Cruz GM, Santos PHLNA, Cortez GG, Rodrigues JAJ (2016) Nickel oxides supported on alumina as oxygen carriers for chemical-looping combustion and reforming of methane. Appl Mech Mater 830:30–37
Barzetti T, Selli E, Moscotti D, Forni L (1996) Pyridine and ammonia as probes for FTIR analysis of solid acid catalysts. J Chem Soc Faraday Trans 92:1401–1407
Brayner R, Bozon-Verduraz F (2003) Niobium pentoxide prepared by soft chemical routes: morphology, structure, defects and quantum size effect. Phys Chem Chem Phys 5:1457–1732
Chun H, Kim D, Lim D, Lee W, Lee H (2010) A synthesis of CO-tolerant Nb2O5-promoted Pt/C catalyst for direct methanol fuel cell; its physical and electrochemical characterization. Int J Hydrog Energy 35:6399–6408
de Diego L, Gayán P, García-Labiano F, Celaya J, Abad A, Adánes J (2005) Impregnated CuO/Al2O3 oxygen carriers for chemical-looping combustion: avoiding fluidized bed agglomeration. Energy Fuel 19:1850–1856
Emeis CA (1993) Determination of integrated molar extinction coefficients for infrared absorption bands of pyridine adsorbed on solid acid catalysts. J Catal 141:347–354
Fiévet F, Brayner R (2013) The polyol process. In: Brayner R, Fiévet F, Cordin T (eds) Nanomaterial: a danger or a promise? Springer-Verlag, London, p 1–25
Gayán P, de Diego L, García-Labiano F, Adánez J, Abad A, Dueso C (2008) Effect of support on reactivity and selectivity of Ni-based oxygen carriers for chemical-looping combustion. Fuel 87:2641–2650
He J, Li Q, Fan Y (2013) Dispersion states and acid properties of SiO2-supported Nb2O5. J Solid State Chem 202:121–127
IUPAC Recommendations (1985) Pure Appl Chem 57:603
IUPAC Recommendations (1994) Pure Appl Chem 66:173
Kitano T, Shishido T, Teramura K, Tanaka T (2012) Brønsted acid property of alumina-supported niobium oxide calcined at high temperatures: characterization by acid-catalyzed reactions and spectroscopic methods. J Phys Chem C 116:11615–11625
Ko E, Weissman J (1990) Structures of niobium pentoxide and their implications on chemical behavior. Catal Today 8:27–36
Kvamsdal M, Jordal K, Bolland O (2007) A quantitative comparison of gas turbine cycles with CO2 capture. Energy 32:10–24
Li J, Lu G, Li K, Wang W (2004) Active Nb2O5-supported nickel and nickel-copper catalysts for methane decomposition to hydrogen and filamentous carbon. J Mol Catal A Chem 221:105–112
Li Y, Yan S, Yang W, Xie Z, Chen Q, Yue B, He H (2005) Effects of support modification on Nb2O5/alpha-Al2O3 catalyst for ethylene oxide hydration. J Mol Catal A Chem 226:285–290
Li S, Xu Q, Uchaker E, Cao X, Cao G (2016) Comparison of amorphous, pseudohexagonal and orthorhombic Nb2O5 for high-rate lithium ion insertion. Crystal Eng Comm 18:2532–2540
Miracca I, Asen KAJ, Coulter C, Curran L, Lowe C, Moure G, Shlasner S (2009) The CO2 capture project (CCP): results from phase II (2004-2009). Energy Procedia 1:55–62
Mozer T, Passos F (2011) Selective CO oxidation on cu promoted Pt/Al2O3 and Pt/Nb2O5 catalysts. Int J Hydrog Energy 36:13369–13378
Nowak I, Ziolek M (1999) Niobium compounds: preparetion, characterization, and application in heterogeneous catalysis. Chem Rev 99:3603–3624
Pröll T, Bolhàr-Nordenkampf J, Kolbitsch P, Hofbauer H (2010) Syngas and a separate nitrogen/argon stream via chemical-looping reforming - a 140kW pilot plant study. Fuel 89:1249–1256
Tanabe K (2003) Catalytic application of niobium compounds. Catal Today 78:65–77
Zafar Q, Mattisson T, Gevert B (2005) Integrated hydrogen and power production with CO2 capture using chemical-looping reforming - redox reactivity of particles of CuO, Mn2O3, NiO, and Fe2O3 using SiO2 as a support. Ind Eng Chem Res 44:3485–3496
Zafar Q, Mattisson T, Gevert B (2006) Redox investigation of transition-state metals Ni, Cu, Fe and Mn supported on SiO2 and MgAl2O4. Energy Fuel 20:34–44
Zhao Y, Zhou X, Ye L, Tsang S (2012) Nanostructured Nb2O5 catalysts. Nano Reviews 3:17631-.17642
Ziolek M (2003) Niobium-containing catalysts - the state of the art. Catal Today 78:47–64
Acknowledgements
Pedro H.L.N.A dos Santos is grateful to Ciências Sem Fronteiras program from CNPq Brazil for financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
dos Santos, P.H.L.N.A., Nowak, S., Lau-Truong, S. et al. Preparation of niobium-based oxygen carriers by polyol-mediated process and application to chemical-looping reforming. J Nanopart Res 21, 89 (2019). https://doi.org/10.1007/s11051-019-4528-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-019-4528-z