
Abstract
A new method to conjugate methoxy polyethylene glycol (mPEG) to C6 position of chitosan under the mild condition is introduced that improves the biocompatibility and water solubility of chitosan. Harsh deprotecting step and several purification cycles are two major disadvantages of the current methods for preparing PEGylated chitosan. In this study, the amine groups at C2 position of chitosan are protected using SDS followed by grafting the PEG. The protecting group of chitosan is simply removed by dialyzing against Tris solution. The chemical structure of the prepared polymer is characterized by FTIR and 1H NMR spectroscopy. Fourier transformed-infrared (FT-IR) and 1H NMR spectra confirmed that the mPEG is successfully grafted to C6 position of chitosan. Prepared methoxy polyethylene glycol (mPEG) is then employed to prepare the nanocapsules for the encapsulation of poor water-soluble drug, propofol. The TEM, AFM, and DLS techniques are used to characterize the prepared nanocapsules size and morphology. The results show a size of about 80 nm with spherical shape for nanocapsules. In vitro drug release is carried out to evaluate the potential of nanocarriers for the intravenous delivery of drugs. The profile of release from formulated nanocapsules is similar to those of commercial lipid emulsion (CLE). In vivo animal sleep-recovery test on rats shows a close similarity between the time of unconsciousness and recovery of righting reflex between nanoparticles and CLE. This study provides an efficient, novel, and easy method for preparing a carrier system that requires less intensive reaction conditions, fewer reaction steps, and less purification steps. In addition, the nanocapsules introduced here could be a promising nano carrier for the delivery of poor water-soluble drugs.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Chitosan is a glycopolymer that has many applications in biotechnology, pharmaceutical, textile, food, cosmetics, and agriculture due to its polycationic structure as well as the nontoxicity, biodegradability, and biocompatibility (Ravi Kumar 2000; Ilium 1998). Unfortunately, chitosan is only soluble in an acidic aqueous solution because of the presence of amine groups. The low-water solubility of chitosan is counted as the major limitation for its application (Jia and Xu 2001). In order to overcome this problem, researchers have attempted to chemically modify chitosan by grafting other polymers such as different molecular weights of mPEG ((Ouchi et al. 1998; Prego et al. 2006; Ganji and Abdekhodaie 2008; Krishna Rao et al. 2012). Due to its hydrophilicity flexibility, non-ionic property, biodegradability, biocompatibility, low toxicity, and immunogenicity (Germershaus et al. 2008), PEG conjugated chitosan has been widely applied in the pharmaceutical fields in the forms of microspheres, nanoparticles, or polymers. This will enhance the stability, prolong half life of active agents, and alter pharmacokinetics, tissue distribution, and pharmacological properties (Greenwald et al. 2003; Chiu et al. 1998; Hu et al. 2008; Du and Hsieh 2007). Even though the modification of chitosan via the amino groups is the most commonly used method in the texts (Sugimoto et al. 1998), some researchers have investigated the PEGylation of chitosan via the hydroxyl groups. The presence of amine groups in the chitosan skeleton makes it an excellent cell transfer agent and provides the possibility of interaction with mucus and mucosal surface (Fiebrig et al. 1995, 1997; Deacon et al. 1999). Modification of hydroxyl group of chitosan with polyethylene glycol) (PEG) was first proposed by Gorochovceva and Makuška (2004). They used Ag2O as catalyst that produced a high degree of PEG substitution on chitosan. They found that, the residual silver salts that remain in the reaction cause the degradation of chitosan during the process of deprotection of phthalic group with hydrazine (Makuška and Gorochovceva 2006). Moreover, after several purification cycles, the silver nanoparticles were not completely removed from the PEGylated chitosan, and the overall yield of reaction was low even after 26–30 h. Malhorta group used NaH as a catalyst which was more promising compared to Ag2O, and the yield of PEGylation reaction could be increased by using low-molecular weight PEG (not less than 1,000). However, NaH used as catalyst makes the reaction conditions alkaline which potentially have a negative effect on phthalic protecting group (Malhotra et al. 2011; Makuška and Gorochovceva 2006). Makuska et al. (2009) (Gruškienė et al. 2009) described three different methods for grafting mPEG onto the hydroxyl group of chitosan in C6 position using three different phthaloyl chitosans such as 6-oxo-2-N-phthaloylchitosan, 6-O-dichlorotriazine-2-N-phthaloyl chitosan, and 3-O-acetyl-2-N-phthaloylchitosan. However, N-Phthaloyl chitosan as a precursor, for the synthesis of PEGylated chitosan, has a drawback associated with the partial destruction of products under protection–deprotection procedure (Makuška and Gorochovceva 2006; Holappa et al. 2004). In addition, because of the reaction conditions, the chitosan backbone is subjected to a significant breakdown during these processes. It has also been described that the deprotection of phthaloyl groups with hydrazine has been found to be incomplete, including up to 20 % phthaloyl groups found in the final product (Lebouc et al. 2005).
In the method that is presented here, the protection of amine groups at C2 position of chitosan was carried out by SDS rather than phthaloyl group which can be simply removed by dialyzing the precipitation against Tris solution. This deprotecting method did not affect the back bone structure of chitosan. In addition, conjugating methoxy polyethylene glycol (mPEG) onto C6 position of chitosan was occurred under a mild condition, required less intensive reaction conditions, fewer reaction steps, and needs less purification steps in short period of time compared to the other previously reported methods (Gruškienė et al. 2009; Liu et al. 2006).
Propofol, chemically named 2,6-diisopropylphenol, is a short-acting intravenously administered hypnotic agent which is used for the induction and maintenance of general anesthesia, sedation for mechanically ventilated adults, and procedural sedation (Miner and Burton 2007). Although propofol has attractive properties as an anesthetic agent, it has low solubility in water that imposes limitation in its injectable formulation (Brambrink et al. 2012). Despite the success of the current formulation of propofol (CLE), numerous disadvantages have been reported for this lipid-based formulation, including instability of lipid-based emulsion (Sundarathiti et al. 2007), hyperlipidemia (Wolf et al. 2001), and pain during injection (Lee et al. 1994), and rapid growth of microorganisms (Wachowski et al. 1999).
In this study, a facile novel method for conjugating methoxy polyethylene glycol (mPEG) onto C6 position of chitosan is introduced. The synthesized polymer is then employed to prepare the nanoparticles for the encapsulation of propofol as poor water-soluble drug model. The transmission electron microscopy (TEM), atomic force microscopy (AFM), and dynamic light scattering (DLS) techniques were used for the characterization of nanocapsules. In addition, in vitro and in vivo propofol release tests were carried out in order to compare the release profile between PEGylated chitosan and CLE.
Materials and methods
Materials
Low-molecular-weight chitosan was obtained from Sigma-Aldrich (USA), having a degree of deacetylation of 80.0 %. Poly(ethylene glycol) monomethyl ether (mPEG) (MW 1000), sodium dodecylsulfate (SDS), sodium hydride (NaH), pyridine, cellulose membrane, acetonitrile, methanol, and glacial acetic acid of analytical grade were obtained from Sigma-Aldrich (Switzerland). Anhydrous tetrahydrofuran (THF), Thiamine pyrophosphate (TPP), anhydrous N,N-dimethylformamide (DMF), sodium hydroxide (NaOH), and oxalyl chloride were obtained from Merck Chemical Co. (Germany). Propofol was purchased from Zhongke Taidou Chemical Co., Ltd. (Shandong, China). Propofol commercial lipid emulsion (CLE) injection was provided from Guorui Pharmaceutical Co. (Sichuan, China).
PEGylation of chitosan
We employed a novel scheme to PEGylate chitosan chemoselectively. The synthesis was consisted of 3 steps: (1) protection of the chitosan amine group with SDS followed by chlorination of protected chitosan, (2) activation of OH- PEG-OCH3 (mPEG) with NaH, and (3) grafting the activated mPEG onto the chlorinated chitosan followed by removing the protecting group (Scheme 2).
Protection and chlorination of chitosan
Deacetylation degree of chitosan was increased according to the method previously described (Torii et al. 2009) (a and b) in Scheme 2. The SDS/chitosan complex is prepared based on the procedure introduced by Cai et al. (2009). Briefly, Sodium dodecylsulfate (SDS)/chitosan complex (SCC) was prepared by mixing acidic solutions of chitosan (2 % w/v) and SDS. The resulting precipitates (C) in Scheme 2 were filtered off, washed three times with distilled water, and freeze-dried to yield a white product. To prepare chlorinated chitosan, 1 g of oxalyl chloride was added to 0.1 g of SCC in 50 mL of pyridine. The reaction was stirred at room temperature for 90 min under nitrogen atmosphere. After 30 min, the reaction was precipitated in ice cold water, filtered, and vacuum dried to yield a chlorinated-SCC, product (D) in Scheme 2.
Activation of mPEG and grafting to chitosan
Methoxy polyethylene glycol (mPEG) was added to a suspension of NaH (10 mg) in 50 mL of anhydrous THF (Table 1). The reaction was stirred at 60 °C for 2 h under nitrogen atmosphere. After 6 h, chlorinated chitosan (60 mg) was added to the reaction mixture and stirred for another 24 h under similar conditions. Finally, the reaction was allowed to cool at room temperature and precipitated in methanol, filtered, washed, and vacuum dried to yield product (E).
Synthesis of PEGylated chitosan
PEGylated-SDS chitosan which was prepared in previous part was added directly to Tris solution. The PEGylated chitosan was precipitated in 15 % Tris(hydroxyl methyl) amino methane aqueous solution (Tris; the pH was adjusted to 8 with 5 M HCl) at room temperature, while protecting group (SDS) was dissolved in Tris solution. The precipitates were collected via centrifugation, washed with 15 % Tris solution and distilled water, and freeze-dried to yield chitosan-grafted-PEG (Scheme 1b). The water-soluble PEGylated chitosan was purified by dialysis against Tris aqueous solution (pH 8) with cellulose membrane (M cut off 10,000) over one week in order to completely remove any toxic residue from the reaction (Cai et al. 2009).

Chemical structure of A Chitosan B PEGylated chitosan
Characterization of PEGylated chitosan
Proton 1H and 13C nuclear magnetic resonance (NMR) were carried out on a 400 MHz Bruker NMR spectrometer using D2O and DMSO as a solvent. Fourier transform infrared spectroscopy (FT-IR) was recorded on a Shimadzu 8400S Spectrometer (Shimadzu 8400S FTIR, Osaka, Japan). A total of 2 % (w/w) of samples was mixed with dry potassium bromide (KBr); the mixture was grounded into a fine powder before being compressed to form KBr disk. Each KBr disk was scanned at resolution of 4 mm/s between 400 and 4,000 cm−1. Thermogravimetric analysis (TGA) was performed on Mettler-Toledo TGA/DSC-(Mettler-Toledo, Leicester, UK) at temperature ranging between 25 and 700 °C at 10 °C/min heating rate under nitrogen atmosphere.
Preparation of chitosan nanocapsules
After deprotection of PEGylated chitosan, the polymer was dissolved in 1 % (v/v) acetic acid solution to yield a concentration of 5 mg/mL, and pH was adjusted to 5. TPP was dissolved in dd H2O to obtain a concentration of 1 mg/mL, and the pH was adjusted to 3 (Malhotra et al. 2009). Nanocapsules were formed after the addition of TPP (drop wise) to PEGylated chitosan solution under constant magnetic stirring for 1 h at room temperature. For preparation of drug encapsulated nanocapsules, predetermined amount of drug (Tables 1, 2) was added to previously prepared PEGylated chitosan solution which was then processed as the above mentioned procedure. For preparing chitosan nanocapsules, predetermined amounts of chitosan were dissolved in 1 % (v/v) acetic acid solution and predetermined amount of drug was added to the solution which was then processed as above.
Brunauer–Emmett–Teller measurement (BET)
Pore size distribution and specific surface area analyses of chitosan and PEGylated chitosan nanocapsules were measured by nitrogen adsorption–desorption using a BELSORPMini II (Japan) at 140 °C. Before the measurement, 150 mg of the nanocapsules were heated at 100 °C for 2 h under vacuum. The specific surface area, pore volume, and average pore diameter of chitosan and PEGylated chitosan nanocapsules were obtained by Brunauer–Emmett–Teller (BET) method using BELSORP analysis software.
Characterization of nanocapsules
Particle size and morphology of nanocapsules were determined by atomic force microscopy (AFM) in the tapping mode transmission electron microscopy (TEM) and dynamic light scattering (DLS). For AFM (JPK, Nanowizard 2, JPK Instruments, Germany), silicon tapping tip (TESP, VEECO) was used with a nominal tip radius less than 10 nm as provided by the manufacturer. Nanocapsules were dispersed in deionized water at 1 mg/mL concentration. A droplet was put on the polyethylene imine coated glass cover slip and air dried. Samples were then imaged using Nano-scope III (Digital Instrument/VEECO) with an E scanner (maximum scan area = 14.2 × 14.2 μm2). The TEM investigation of nanocapsules was carried out the following negative staining with uranyl acetate. Briefly, a drop of the nanocapsules suspension (same concentration as AFM test) was placed on formvar-coated copper grids (Ted Pella, Inc., Redding, Ca) and allowed to equilibrate. The excess amount of liquid was detached with filter paper, and a drop of 2 % W/V uranyl acetate was mixed to the grid and air dried after 3 min followed by TEM examination (TEM, Philips/FEI, Inc., NY). The diameters of at least fifty individual nanocapsules in the TEM images were determined manually using a digital caliper which was used to calculate the average particle size. For DLS measurement, a Malvern Zetasizer Nano ZS (Malvern instruments, UK) was used. The polymer particles were suspended in dd H2O and measured at a fixed scattering angle of 90°.
Determination of drug loading and encapsulation efficiency
In order to determine the drug loading, LC (w/w %) of PEGylated chitosan polymer, nanocapsules were freeze-dried and dissolved in methanol, and the drug content of the nanocapsules was determined on a Agilent Technologies HPLC system (Agilent Technologies Inc, 1200, California, USA) equipped with a UV detector (Agilent Technologies Inc, 1200) and reversed phase column (ODS C18, 5 μm, 4.6 × 250 mm2, Dikma, China). The mobile phase consisted of 80–20 Acetonitrile–water (v/v %), respectively. The mobile phase was pumped at a flow rate of 1.0 mL/min, and propofol concentration was measured at 270 nm. The LC of the nanocapsules was then calculated according to the following formula:
In order to determine the concentration of drug in the aqueous phase of nanocapsules and commercial propofol emulsion, two phases were separated using ultracentrifuge at 10,000 rpm (16000 g) for at least 30 min. Amicon ultra centrifugal filter units (Amicon ultra 4 k, Milipore, Usa) were filled with 350 μl of propofol sample, and after centrifugation, the separated aqueous phase was analyzed by HPLC. Encapsulation efficiency (EE) of nanocapsules was calculated according to the following formula:
In vitro drug release
In vitro release of encapsulated propofol from the nanocapsules was determined under sinks conditions (volume of release medium used was enough to dissolve at least five times the quantity of drug loaded in nanocapsules). 5 mg of nanocapsules was dispersed in 5 mL of phosphate-buffer saline (PBS, pH 7.4) and placed into a dialysis bag with molecular weight cut off of 3,000 D (Sigma, Germany). The dialysis bag was placed in a flask containing 30 mL of release medium (PBS, pH 7.4) containing 30 % (v/v) alcohol (sink condition). The whole assembly was shaken at 100 rpm and 37 °C. At predetermined time intervals, 5 mL of the release medium was removed and replaced with the fresh medium. The content of propofol in the medium was measured by HPLC as described previously. The cumulative release percentage of propofol was calculated and compared with the CLE used as control.
In vivo drug release
Male SD rats at the beginning of the experiments were provided with standard food and tap water, and exposed to alternating 12 h periods of light and darkness. Temperature and relative humidity were maintained at 25 °C and 50 %, respectively. After 7 days of adaptation period, the rats were fasted for 12 h, but allowed freely to access water before the experiments. Eighteen animals were randomly divided into two groups (n = 9) using a random number generator. The prepared nanocapsules and CLE were injected intravenously (nanocapsules sterilized by filtration through a 200-nm pore filter) via the caudal vein of rats in each group at a single dose of 10 mg/kg. The end of injection was consummated as time zero (t = 0). After each injection, the time to loss of righting reflex was recorded for each animal. Rats were maintained in dorsal or lateral recumbency during evaluation, and the time to regain locomotor activity and righting reflex return were recorded.
Result and discussion
Synthesis and characterization of mPEG-grafted chitosan
The deacetylated chitosan was characterized by 13C nuclear magnetic resonance (NMR). The NMR spectra of the native chitosan and deacetylated chitosan are reported in Table 3. In the NMR spectra of the native chitosan, the presence of two bands of 23.88 and 175.04 ppm are ascribed to the CH3 and C=O component, respectively. The FTIR spectra of the native and the SDS/chitosan samples are shown in Fig. 1a, b. In the FTIR spectra of the native chitosan, the presence of broad band at 3,200–3,600 cm−1 is ascribed to the OH and NH component in chitosan (Kulkarni et al. 2005). This peak is also present in the SDS/chitosan spectra; however, the reduction in the peak size accompanied by appearance of new bands at 1,180 (S–O) and 810 (C–O–S) cm−1 suggests that the attachment of the sulfate group of SDS took place on glucosamine groups. Moreover, the magnitude of the peak at 3,200–3,600 cm−1, which can be attributed to the hydrogen-bonds of the hydroxyl and NH groups, has been decreased in the modified sample compared to the native chitosan. This observation can be attributed to the addition of SDS to chitosan with interaction between the sulfate group of SDS and amine group of glucosamine in chitosan.

Fourier transform infrared spectra of the chitosan, SDS/chitosan, mPEG, PEGylated SDS/chitosan, and PEGylated chitosan
The 1H NMR and 13C NMR spectroscopy were used to gather more information about the SDS/chitosan complex (Table 3). The sulfate/amino group ratio in SDS/chitosan was measured according to the integrated area ratio between peaks at 0.85 ppm (CH3 in SDS) and 2.85 ppm (H2 of unsubstituted glucosamine in chitosan) (Gruškienė et al. 2009). The sulfate/amino group ratio in the SDS/chitosan complex was calculated to be 1:1 which is close to the stoichiometric ratio indicating the complete complexation of SDS/chitosan. When the sulfate/amino group ratio in the complex goes above 0.6, the complex was easily dissolved in pyridine. The ionic strength of the medium has a significant effect on the electrostatic interaction between the surfactant and chitosan. We found that SDS could be soluble in Tris aqueous solution at any concentration. Thus, we used Tris aqueous solution for deprotecting SDS from SDS/chitosan. The SDS/chitosan complex could be decomposed to SDS and chitosan simply by precipitation in Tris aqueous solution (pH 8.0). Viscometry investigation showed that the deprotected chitosan has the same molecular weight (M n ) as the initial chitosan which confirmed that the addition and removal process had no effect on the chitosan structure which indicated that our procedure was not affect the back bone of chitosan rather than using phthaloyl as protecting group (Dembo and Starodoubtsev 2001).
The synthesis reaction and process of chitosan-PEG is illustrated schematically in Scheme 2. In order to increase the rate of reaction between PEG and chitosan, chitosan was chlorinated before addition to mPEG, and then activated PEG was added to chlorinated chitosan. The hydroxyl group of mPEG was activated using NaH, and then coupled to the chlorinated group of chitosan, forming a graft copolymer of mPEG-chitosan. The structures of chitosan-PEG derivatives were confirmed by FTIR and 1H NMR analyses. The FTIR spectrum of native chitosan, chlorinated chitosan, and chitosan-PEG are shown in Fig. 1a, b, d, respectively. In the FTIR spectrum of native chitosan, the presence of broadened peak at 3,200–3,600 cm−1 was attributed to –OH and –NH stretching vibration in which the –OH stretching vibration was overlapped by the –NH stretching vibration (Fig. 1a). The peak at 1,640 cm−1 referred to the amide band of chitosan, the peak at 1,550 cm−1 corresponded to the symmetrical stretch vibration absorption of the amino group, and the band at 1,381 cm−1 referred to stretching vibration of C–N group. In the FTIR spectrum, the presence of a band at 1,024 cm−1 is ascribed to the stretch vibrations of C–O in chitosan (Peng et al. 2010). In the FTIR spectra of the chitosan-PEG, the presence of the band corresponding to the hydroxyl group of chitosan slightly shifted which is attributed to PEGylation of the hydroxyl group. However, the reduction in the peaks size, accompanied by appearance of new bands at 1,024 (C–O), 1,280, 1,320, 1,480, 810, and 3,200–3,600 (–CH2–O–CH2) suggested that the reaction took place on some of the hydroxyl groups of chitosan when compared to Fig. 1c that represents the FTIR spectrum of native mPEG with the characteristic peaks at 2,890 cm−1 (C–H stretching), 1,100 (C–O stretching), 1,474, and 1,278 cm−1. Thus, it is supposed that mPEG was successfully attached to chitosan via the NaH activation system.

The process of chitosan PEGylation A Native chitosan B Deacetylated chitosan C SDS/chitosan complex D Chlorinated SDS/chitosan complex E PEGylated SDS/chitosan F PEGylated chitosan
The 1H NMR spectrum of the chitosan-PEG showed the sharp peak of PEG-OCH3 at 3.31 ppm, strong broad signal at 3.2–3.8 ppm (PEG–OCH2CH2O–) which were not separated due to the overlapping of a more intense PEG peak with the peak from chitosan pyranose ring (Du and Hsieh 2007).The peak at 2.85 ppm is always referred to as the H-2 of the unsubstituted glucosamine residue in the chitosan (Sashiwa et al. 2002). The remaining of the attached SDS to chitosan was calculated according to the integration area ratios of A 1.85/A 2.85 in all the chitosan-SDS-PEG samples. We found that the A 1.85/A 2.85 ratio is almost the same as those in the SDS/chitosan complex before reaction with PEG, which suggests that the salt bridge between chitosan and SDS was steady enough, and all amino groups of chitosan were protected by complexation between SDS and chitosan during the addition of PEG to chitosan.
The SDS was removed from the chitosan-PEG complex, easily by dialyzing the solution against tris aqueous solution for one week. The chemical structure of dialyzed polymer was confirmed by FTIR and 1H NMR analyses. The FTIR spectrum of the precipitated polymer verified the absence of peaks at 1,280 and 810 cm−1 assigned to the sulfate group of SDS. The signals at 0.85, 1.22, 1.46, and 3.70 ppm which correspond to the SDS were all moved out, indicating that SDS was removed from chitosan-PEG. The degree of PEGylation was determined from the 1H NMR spectrum by integration of area ratio between the peaks of NHAc at 1.85 ppm of chitosan and -OCH3 of mPEG at 3.3.
Figure 2 represented the TGA and DTA curves of chitosan and PEGylated chitosan. The major weight loss of chitosan was occurred between 187 and 476 °C; however, the main weight loss of chitosan-PEG was occurred between 179 and 500 °C which is close to the decomposition temperature of mPEG (177 °C). The slight shift to lower temperature could be attributed to grafting of mPEG to chitosan. In addition, DTG data point out that mPEG was completely decomposed at 400 °C. Thus, it could be concluded that the mPEG was conjugated to the hydroxyl group of chitosan via “grafting to” method (Fig. 2).

TGA and DTG spectra of the chitosan and PEGylated chitosan
Nanocapsules characterization
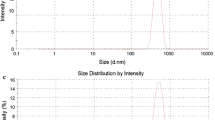
The particle size measurement is commonly used to characterize nanocapsules in order to study their dispersion and aggregation process. In addition, particle size affects biological handling of nanocapsules (Desai et al. 1997), and the sub-hundred nanometer particle size is helpful in the drug delivery system, since they have shown a higher cellular and tissue uptake in this size range (Hillyer and Albrecht 2001; Brooking et al. 2001; Florence et al. 1995). Table 4 shows the zeta potential of chitosan-PEG nanocapsules, chitosan, and commercial emulsion of propofol (CLE). In addition, the particles size distribution spectrum for the chitosan-PEG nanocapsules is represented in Fig. 3 that shows the average particle size of 80.6 ± 1.2 nm. Propofol emulsion and other lipid emulsions for intravenous injection are usually prepared with oil droplets raging from 150−300 nm in size (Han et al. 2001). We found that the average particle size of CLE was 230.7 ± 3.5 nm, and no droplet more than 1 μ was detected. The transparency of the microemulsion or emulsion solutions is directly correlated to the size distribution of the particles. This means that particles with less than one-quarter of the average wavelength of visible light (0.560 m) allow light to pass through the solution which results in a clear appearance (Baker and Naguib 2005). The propofol-loaded chitosan-PEG nanocapsules prepared in this study present a clear appearance with an average particle size of around 80 ± 1.2 nm, whereas the propofol commercial emulsion is a white milky solution due to the presence of much larger droplets (Fig. 3). The milky, opaque appearance of propofol emulsion would prevent detection of macro particles which is considered as a major disadvantage for a solution that is intended for intravenous injection. Finkelstein et al. (1990) have found particles with the approximate dimension of 2.5–17 mm in CLE. These types of visible matter should be checked after manufacturing the injectables and before the injection. However, the opaque appearance of the emulsion makes it impossible to monitor such macrocapsules which could impose a potential danger to patients. The propofol nanocapsules fabricated in the present study is completely transparent which allows the inspection of such visible macroparticles and provide a much safer injection for the patients.

The effective diameter and size distribution of propofol encapsulated nanocapsules measured by particle size analyzer and representation of propofol-loaded nanocapsules which shows a clear solution
Nanocapsules in the lateral and vertical dimension were analyzed by a sectional analysis tool of Nanoscope 5.12b.48 software. Figure 4 shows the AFM images of nanocapsules that revealed an average diameter in the lateral dimension of 40 nm ± 10 nm and vertical diameter of 23.7 nm ± 10 nm. Figure 4 also shows nanocapsule shapes which is in agreement with the TEM images (Fig. 5) presenting a spherical morphology. The difference in average particle size measured by DLS might be due to the swelling of nanocapsules in solution during the measurement.

Tapping mode AFM images of a nanocapsules in air b 3D images of nanocapsules in a 400 × 400 nm2 scan area

TEM image of PEG-chitosan nanocapsules that negative stained with 2 % w/v uranyl acetate
In vitro drug release
Drug loading and encapsulation efficiency of the chitosan-PEG is represented in Table 4 that shows a dependency to the chitosan-PEG concentration. Increasing the concentration of chitosan-PEG from 0.1 to 1 % w/v in the formulation of nanocapsules caused an increase in propofol encapsulation from 48 to 99 % (Table 4). However, because of low drug loading of chitosan nanocapsules, the investigations were carried out on CLE as control rather than chitosan nanocapsules. In addition, drug encapsulation and encapsulation efficiency in nanocapsules were dependent on the amount of drug added to the formulation as represented in (Table 2). Furthermore, the effect of PEGylation degree on the encapsulation efficiency was investigated in Table 1. The encapsulation efficiency of PEGylated chitosan improved with increasing the degree of PEGylation from 0.12 to 0.64 in the formulation of nanocapsules which caused an increase in drug encapsulation from 41 to 99 % (Table 1). We found a non-linear relation between the amount of drug added to the formulation and encapsulation efficiency. The encapsulation efficiency was 99 % by addition of 1 % v/v of propofol, whereas the encapsulation efficiency decreased to 43 % when 3 % of propofol was used. This could be explained according to the contribution of hydrophobic–hydrophobic interaction between propofol and the hydrophobic group (CH3 in mPEG) of chitosan-PEG. Increasing the concentration of chitosan-PEG is expected to increase the number of hydrophobic groups available for interaction with the hydrophobic drug (propofol), resulting in a better drug entrapment in the nanocapsules. Due to the limited number of hydrophobic groups in chitosan-PEG nanocapsules which are available for interaction with the drug, an increase in the drug concentration has resulted in a decrease in drug encapsulation efficiency. Drug loading and encapsulation efficiency of the chitosan-PEG are represented in Table 4 that shows dependency to the chitosan-PEG concentration.
The specific surface area, pore volume, and average pore diameter of nanocapsules were measured by BET method using BELSORP analysis software. The adsorption isotherm obtained has been analyzed using BET equation and BJH algorithm. The specific surface area (m2/g), average pore diameter (nm), and specific pore volume (cm3/g) of chitosan were 30.79 (m2/g), 0.65 (nm), 0.38 (cm3/g), respectively, whereas those for PEGylated chitosan were 30.19 (m2/g),0.85 (nm), and 0.35 (cm3/g), respectively. As a result, the specific surface areas, the specific pore volumes, and average pore diameters have shown that chitosan nanocapsules consist of smaller pore size and more specific surface area rather than PEGylated chitosan. This correlated to the addition of PEG branches to the chitosan backbone which limited the interactions between the polymeric chains of chitosan.
The ability of chitosan-PEG nanocapsules to deliver propofol was measured by drug release rate analysis. Commercial propofol emulsion (CLE) was used as control (Fig. 6). Figure 6 shows that 20 % of propofol was released from nanocapsules during first 2 h, and about 72 % of propofol was released after 48 h. In addition, no burst release was observed for samples and controls during the initial period of investigation. The drug release profile of chitosan-PEG nanocapsules was generally similar to the commercial propofol emulsion (CLE) (Chung et al. 2001; Hung et al. 2007; Kandadi et al. 2011; Li et al. 2011).

In vitro release of propofol from encapsulated nanocapsules and commercial lipid emulsions (CLE) as control (Mean ± SD, n = 5)
In vivo drug release
The average time to loss and recovery of righting reflex was investigated for encapsulated nanocapsules and CLE. The animals righting reflex is quickly vanished (during 35 s of dose administration) after administration of both propofol formulations. Figure 7 presents the results of recovery of the righting reflex indicating the recovery time of 7.21 ± 3.7 and 7.25 ± 2.5 min for encapsulated nanocapsules and CLE, respectively. It was found that both groups of animals which were administrated by propofol loss and recover their righting reflex after about 7.2 min for both formulations. Furthermore, no sign of toxic effects of usage was observed either from nanocapsules formulation or CLE which indicates that nanocapsules formulation has similar pharmacological effects as CLE. It is believed that small difference in propofol release profile observed between nanocapsules formulation and CLE could be related to the variation in their influence on the central nervous system of rats leading to similar pharmacological effects.

Sleep-recovery investigation of nanocapsules and CLE as control. (Onset of sleep was less than 35 Sec)
Conclusion
In this study, we developed a facile method for preparation of methoxy poly (ethylene glycol) (mPEG)-grafted chitosan to improve the biocompatibility and water solubility of chitosan. The drug encapsulated nanocapsules showed a particle size of 80 nm which is smaller and more uniform than commercial lipid emulsion (CLE). The profile of release from formulated nanocapsules was similar to those of commercial lipid emulsion (CLE). In vivo animal sleep-recovery test on rates showed a close similarity in time of unconsciousness and recovery of righting reflex between nanoparticles and CLE. The results showed that prepared nanocapsules could be a promising system for delivery of poor water-soluble drugs such as propofol. We expect that the antibacterial property of chitosan could prevent microbial growth in formulated solution which would be a vital advantage over commercial lipid emulsion (CLE). This study provided a new and simple method for fabrication of a nanocarrier system for delivery of hydrophobic drugs at the same time as overcoming some of the drawbacks of CLE. Future studies should be directed toward safety evaluation of PEGylated nanocapsules for clinical application.
References
Baker MT, Naguib M (2005) Propofol: the challenges of formulation. Anesthesiology 103(4):860–876
Brambrink AM, Dissen GA, Martin LD, Creeley CE, Olney JW (2012) Propofol-induced apoptosis of neurons and oligodendrocytes in neonatal macaque brain. Am Soc Anesthesiol
Brooking J, Davis S, Illum L (2001) Transport of nanoparticles across the rat nasal mucosa. J Drug Target 9(4):267–279
Cai G, Jiang H, Tu K, Wang L, Zhu K (2009) A facile route for regioselective conjugation of organo-soluble polymers onto chitosan. Macromol Biosci 9(3):256–261
Chiu H-C, Chern C-S, Lee C-K, Chang H-F (1998) Synthesis and characterization of amphiphilic poly (ethylene glycol) graft copolymers and their potential application as drug carriers. Polymer 39(8):1609–1616
Chung H, Kim TW, Kwon M, Kwon IC, Jeong SY (2001) Oil components modulate physical characteristics and function of the natural oil emulsions as drug or gene delivery system. J Control Release 71(3):339–350
Deacon M, Davis S, White R, Nordman H, Carlstedt I, Errington N, Rowe A, Harding S (1999) Are chitosan–mucin interactions specific to different regions of the stomach? Velocity ultracentrifugation offers a clue. Carbohydr Polym 38(3):235–238
Dembo A, Starodoubtsev S (2001) Interaction between polyelectrolyte gel-surfactant complexes with oppositely charged polymer and surfactant components. Macromolecules 34(8):2635–2640
Desai MP, Labhasetwar V, Walter E, Levy RJ, Amidon GL (1997) The mechanism of uptake of biodegradable microparticles in Caco-2 cells is size dependent. Pharm Res 14(11):1568–1573
Du J, Hsieh Y-L (2007) PEGylation of chitosan for improved solubility and fiber formation via electrospinning. Cellulose 14(6):543–552
Fiebrig I, Harding SE, Rowe AJ, Hyman SC, Davis SS (1995) Transmission electron microscopy studies on pig gastric mucin and its interactions with chitosan. Carbohydr Polym 28(3):239–244
Fiebrig I, Vårum KM, Harding SE, Davis SS, Stokke BT (1997) Colloidal gold and colloidal gold labelled wheat germ agglutinin as molecular probes for identification in mucin/chitosan complexes. Carbohydr Polym 33(2):91–99
Finkelstein A, Lokhandwala BS, Pandey NS (1990) Particulate contamination of an intact glass ampule. Anesthesiology 73(2):362–363
Florence A, Hillery A, Hussain N, Jani P (1995) Factors affecting the oral uptake and translocation of polystyrene nanoparticles: histological and analytical evidence. J Drug Target 3(1):65–70
Ganji F, Abdekhodaie M (2008) Synthesis and characterization of a new thermosensitive chitosan–PEG diblock copolymer. Carbohydr Polym 74(3):435–441
Germershaus O, Mao S, Sitterberg J, Bakowsky U, Kissel T (2008) Gene delivery using chitosan, trimethyl chitosan or polyethylenglycol-graft-trimethyl chitosan block copolymers: establishment of structure–activity relationships in vitro. J Control Release 125(2):145–154
Gorochovceva N, Makuška R (2004) Synthesis and study of water-soluble chitosan-O-poly (ethylene glycol) graft copolymers. Eur Polymer J 40(4):685–691
Greenwald RB, Choe YH, McGuire J, Conover CD (2003) Effective drug delivery by PEGylated drug conjugates. Adv Drug Deliv Rev 55(2):217–250
Gruškienė R, Čiuta G, Makuška R (2009) Grafting of poly (ethylene glycol) to chitosan at c (6) position of glucosamine units via “click chemistry” reactions. Chemija 20(4):241–249
Han J, Davis SS, Washington C (2001) Physical properties and stability of two emulsion formulations of propofol. Int J Pharm 215(1):207–220
Hillyer JF, Albrecht RM (2001) Gastrointestinal persorption and tissue distribution of differently sized colloidal gold nanoparticles. J Pharm Sci 90(12):1927–1936
Holappa J, Nevalainen T, Savolainen J, Soininen P, Elomaa M, Safin R, Suvanto S, Pakkanen T, Másson M, Loftsson T (2004) Synthesis and characterization of chitosan N-betainates having various degrees of substitution. Macromolecules 37(8):2784–2789
Hu F-Q, Meng P, Dai Y-Q, Du Y-Z, You J, Wei X-H, Yuan H (2008) PEGylated chitosan-based polymer micelle as an intracellular delivery carrier for anti-tumor targeting therapy. Eur J Pharm Biopharm 70(3):749–757
Hung C-F, Fang C-L, Liao M-H, Fang J-Y (2007) The effect of oil components on the physicochemical properties and drug delivery of emulsions: tocol emulsion versus lipid emulsion. Int J Pharm 335(1):193–202
Ilium L (1998) Chitosan and its use as a pharmaceutical excipient. Pharm Res 15(9):1326–1331
Jia Z, Xu W (2001) Synthesis and antibacterial activities of quaternary ammonium salt of chitosan. Carbohydr Res 333(1):1–6
Kandadi P, Syed MA, Goparaboina S, Veerabrahma K (2011) Brain specific delivery of pegylated indinavir submicron lipid emulsions. Eur J Pharm Sci 42(4):423–432
Krishna Rao K, Ramasubba Reddy P, Lee Y-I, Kim C (2012) Synthesis and characterization of chitosan–PEG–Ag nanocomposites for antimicrobial application. Carbohydr Polym 87(1):920–925
Kulkarni AR, Hukkeri VI, Sung HW, Liang HF (2005) A Novel Method for the Synthesis of the PEG-Crosslinked Chitosan with a pH-Independent Swelling Behavior. Macromol Biosci 5(10):925–928
Lebouc F, Dez I, Desbrières J, Picton L, Madec P-J (2005) Different ways for grafting ester derivatives of poly (ethylene glycol) onto chitosan: related characteristics and potential properties. Polymer 46(3):639–651
Lee T, Loewenthal A, Strachan J, Todd B (1994) Pain during injection of propofol. Anaesthesia 49(9):817–818
Li X, Zhang Y, Fan Y, Zhou Y, Wang X, Fan C, Liu Y, Zhang Q (2011) Preparation and evaluation of novel mixed micelles as nanocarriers for intravenous delivery of propofol. Nanoscale Res Lett 6(1):1–9
Liu L, Li F, Ye Fang, Guo S (2006) Regioselective grafting of poly (ethylene glycol) onto chitosan and the properties of the resulting copolymers. Macromol Biosci 6(10):855–861
Makuška R, Gorochovceva N (2006) Regioselective grafting of poly (ethylene glycol) onto chitosan through C-6 position of glucosamine units. Carbohydr Polym 64(2):319–327
Malhotra M, Kulamarva A, Sebak S, Paul A, Bhathena J, Mirzaei M, Prakash S (2009) Ultrafine chitosan nanoparticles as an efficient nucleic acid delivery system targeting neuronal cells. Drug Dev Ind Pharm 35(6):719–726
Malhotra M, Lane C, Tomaro-Duchesneau C, Saha S, Prakash S (2011) A novel method for synthesizing PEGylated chitosan nanoparticles: strategy, preparation, and in vitro analysis. Int J Nanomed 6:485
Miner JR, Burton JH (2007) Clinical practice advisory: emergency department procedural sedation with propofol. Ann Emerg Med 50(2):182–187
Ouchi T, Nishizawa H, Ohya Y (1998) Aggregation phenomenon of PEG-grafted chitosan in aqueous solution. Polymer 39(21):5171–5175
Peng H, Xiong H, Li J, Chen L, Zhao Q (2010) Methoxy poly (ethylene glycol)-grafted-chitosan based microcapsules: synthesis, characterization and properties as a potential hydrophilic wall material for stabilization and controlled release of algal oil. J Food Eng 101(1):113–119
Prego C, Torres D, Fernandez-Megia E, Novoa-Carballal R, Quinoa E, Alonso M (2006) Chitosan–PEG nanocapsules as new carriers for oral peptide delivery: effect of chitosan pegylation degree. J Control Release 111(3):299–308
Ravi Kumar MN (2000) A review of chitin and chitosan applications. React Funct Polym 46(1):1–27
Sashiwa H, Kawasaki N, Nakayama A, Muraki E, Yamamoto N, Aiba S-I (2002) Chemical modification of chitosan. 14: 1 Synthesis of water-soluble chitosan derivatives by simple acetylation. Biomacromolecules 3(5):1126–1128
Sugimoto M, Morimoto M, Sashiwa H, Saimoto H, Shigemasa Y (1998) Preparation and characterization of water-soluble chitin and chitosan derivatives. Carbohydr Polym 36(1):49–59
Sundarathiti P, Boonthom N, Chalacheewa T, Jommaroeng P, Rungsithiwan W (2007) A comparison of propofol-LCT with propofol-LCT/MCT on pain of injection. Med J Med Assoc Thail 90(12):2683
Torii Y, Ikeda H, Shimojoh M, Kurita K (2009) Chemoselective protection of chitosan by dichlorophthaloylation: preparation of a key intermediate for chemical modifications. Polym Bull 62(6):749–759
Wachowski I, Jolly DT, Hrazdil J, Galbraith JC, Greacen M, Clanachan AS (1999) The growth of microorganisms in propofol and mixtures of propofol and lidocaine. Anesth Analg 88(1):209–212
Wolf A, Weir P, Segar P, Stone J, Shield J (2001) Impaired fatty acid oxidation in propofol infusion syndrome. Lancet 357(9256):606–607
Acknowledgments
The authors wish to express their gratitude to Iran National Science Foundation (INSF) and Amirkabir University of Technology for their support in carrying out this project. We would also like to thank Prof. A. Khademhosseini (Hravard medical school), Ms. F. Khosravi, and Dr. Ramin Omidvar in nano lab of Amirkabir University of Technology for their help.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Najafabadi, A.H., Abdouss, M. & Faghihi, S. Preparation and characterization of PEGylated chitosan nanocapsules as a carrier for pharmaceutical application. J Nanopart Res 16, 2312 (2014). https://doi.org/10.1007/s11051-014-2312-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-014-2312-7