
Abstract
Magnetic nanoparticles with immobilized metal ligands were prepared for the separation of antibody fragments. First, iron oxide nanoparticles were produced in a solvothermal synthesis using triethylene glycol as solvent and iron(III) acetylacetonate as organic precursor. Via functionalization of the particles with priorly reacted 3-glycidoxypropyltrimethoxysilane and N α,N α-bis(carboxymethyl)-l-lysine (NTA), and charging with Ni2+, magnetic affinity adsorbents were obtained. The particles were applied to separate a His-tagged antibody fragment from a heterogeneous protein mixture of a microbial cultivation supernatant. Binding properties and specificity for purification of the target product ABF D1.3 scFv were optimized regarding the GNTA concentration and were found superior as compared to commercially available systems. A molar ratio of 1:2 Fe2O3:GNTA was most beneficial for the specific purification of the antibody fragment.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Antibodies and antibody fragments are important products for therapy and diagnostics (Frenzel et al. 2012). Their growing demand increases the pressure on reducing process costs and time-to-market development times. A key to efficient production is the straightforward purification of these high-value products. This down-stream processing is the most expensive part and might comprise 50–80 % of the whole process costs (Roque et al. 2004). Today, antibodies are commonly isolated by protein-A chromatography (Low et al. 2007), which is complex to be implemented into continuous processes (Cristancho et al. 2013) and exhibits limitations in scale and process integrity. Therefore, an alternative system to reduce costs and process complexity is most desirable. In this report, we present an easy method to functionalize superparamagnetic iron oxide particles to achieve a selective system with high-binding capacity for protein purification. Magnetic nanoparticles are investigated extensively for potential biomedical applications (Mornet et al. 2004; Duguet et al. 2006; Ghosh Chaudhuri and Paria 2011). Due to their distinct physical and chemical properties, functionalized magnetic nanoparticles are highly suitable for the specific separation of a target protein from a heterogeneous mixture (Sopaci et al. 2009; Lee et al. 2012; Chang et al. 2008; Faraji et al. 2010; Xu et al. 2004). To serve as a potential alternative in integrated continuous protein removal (Käppler et al. 2009; Bucak et al. 2003), it is essential that the particles are highly stable, do not agglomerate, are biocompatible, and reusable. One method pursued to meet this aim is the encapsulation of the particles into a polymeric matrix obtaining a bead-like system (Palecek and Fojta 2007; Csetneki et al. 2004). Nonetheless this encapsulation leads to a decrease of the magnetic response (Arias et al. 2007; Gupta and Gupta 2005) and the particle diameter increases considerably, corresponding to a decrease in the specific surface area. Another strategy is the use of plain nanoparticles functionalized with small molecules like nitrilotriacetic acid or iminodiacetic acid and charging them with metal ions like Ni2+, Zn2+, or Co2+. This enables the immobilization of histidine-tagged proteins directly out of the protein mixture (Hainfeld et al. 1999; Gu et al. 2006). So far, the synthesis and functionalization of magnetic particles in the nanometer scale that exhibit narrow size distributions with high specific surface area and a reproducible functionalization pattern is challenging (Heyd et al. 2011). Coprecipitation is the most frequently applied method to synthesize the magnetic nanoparticles. Via this route it is possible to obtain large quantities of nanoparticles; nonetheless, the control over size, shape, and crystallinity of the particles is very limited. Lee et al. (2006) used imidazole-stabilized Ni/NiO nanoparticles to magnetically separate the His-tagged proteins. Shieh et al. (2006) present an aqueous phase synthesis of magnetic nanoparticles coated with NH +3 , functionalizing them further with glutaraldehyde and N α,N α-bis(carboxymethyl)-l-lysine (NTA).
Here, we want to present a facile and scalable approach as an alternative path. Homogeneous, highly crystalline Fe2O3 nanoparticles with a large specific surface area and a narrow size distribution are obtained via the solvothermal synthesis using triethylene glycol (TEG) as high-boiling, environmentally compatible solvent. The individual nanoparticles are further functionalized with 3-glycidoxypropyltrimethoxysilane (GLYMO) and NTA coupled in a previous step, allowing direct charging of the particles with Ni2+ ions and the selective separation of antibody fragments with high-binding capacity.
Experimental
Synthesis of magnetic nanoparticles
Ethyl acetate, TEG, iron(III) acetylacetonate (Fe(acac)3, ≥99 %), 3-glycidoxypropyltrimethoxysilane (GLYMO, ≥98 %), NTA (≥97 %), and nickel(II) sulfate ≥99 % (all obtained from Sigma-Aldrich) were used without further purification. In a typical procedure, a 1.5 L reactor (Polyclave, type 3/1, Büchi Glas Uster) was filled with 50 g Fe(acac)3 and 1 L solvent (Grabs et al. 2012).
Preparation of GLYMO + NTA (GNTA)
The silane was prepared as described by Anspach (1994) with little modifications. Briefly, 2.82 g NTA was dissolved in 50 mL H2O dest. with 14.13 mL NaOH 10 M. The solution was left to cool to 273 K in an ice bath. Subsequently, 2.375 mL GLYMO was added drop wise to the solution at 273 K whilst stirring vigorously. The reaction mixture was allowed to warm to room temperature and maintained at this temperature for 4 h. Finally, the temperature was raised to 338 K and the mixture was stirred overnight. A stable solution with no precipitation or polymerization of the silane was obtained. IR: 3578 cm−1 (–OH); 2854 cm−1 (–OH); 2360 cm−1 (Si–C); 1592 cm−1 (–NH–); 1444 cm−1 (–CH 2–/–CH 3); 1410 cm−1 (–OH); 1334 cm−1 (–OH); 1092 cm−1 (Si–O–Si); 878 cm−1 (Si–OH); 811 cm−1 (Si–O); 776 cm−1 (Si–O); 611 cm−1 (COOH) (see S.I., Fig. 0.1).
Coupling of GNTA with particles
In a typical procedure, 10 mL of the stable particle dispersion in TEG (100 mg Fe2O3) was precipitated with ethyl acetate and washed twice with ethyl acetate, separating the particles by using a permanent magnet, finally redispersing the particles in 10 mL deionized water. A quantity of 0.1 g Fe2O3 particles in water was added to variable amounts of the GNTA solution, adjusting the pH with HCl 3 M to 11. The temperature of the suspension was raised to 368 K and kept for 3 h. Subsequently, the surface-modified Fe2O3 nanoparticles were precipitated with a 50/50 (v/v) ethyl acetate/ethanol mixture and washed twice with ethyl acetate. Finally, the particles were redispersed in 10 mL deionized water.
Charging the surface-modified particles with Ni2+-ions
A volume of 10 mL Fe2O3–GNTA dispersion containing 0.1 g of particles was mixed with 0.169 g NiSO4 previously dissolved in 2 mL water. Then the mixture was shaken for 24 h at room temperature to reach equilibrium. Subsequently, the particles were separated with a permanent magnet and washed twice with 10 mL deionized water, redispersing them finally in 10 mL of water, resulting in the removal of the excess unbound Ni2+ metal ions.
Purification of antibody fragment (ABF) D1.3 scFv
GNTA-Ni2+-functionalized magnetic nanoparticles were used to selectively purify His-tagged ABF D1.3 scFv from a bacterial cultivation supernatant. It was derived from a fed-batch cultivation of Bacillus megaterium YYBm1 cells as previously published (David et al. 2011). Residual cells were removed by centrifugation and sterile filtration (Sartorius, Minisart syringe filters, 0.2 μm). The nanoparticles were added to 7.5 mL supernatant to reach a concentration of 0.24 mgNP/mL and incubated for 20 min at room temperature. After magnetic removal of the particles, they were resuspended in 100 μL binding buffer (40.1 M Hepes, 1.6 M NaCl). A 50 μL aliquot was used for further analysis of non-washed particles. The remaining 50 μL was washed three times with 1 mL binding buffer. After magnetically removing the particles, they were vacuum dried at 40 °C and subsequently resuspended in 50 μL binding buffer. For SDS-PAGE analysis the 50 μL aliquots, taken before and after washing, respectively, were mixed with 17 μL SDS sample buffer (fourfold) and heated for 3 min at 90 °C. In each SDS-PAGE lane 17 μL was analyzed. Densitometric analysis was performed with the open source software ImageJ (Schneider et al. 2012). The purity of the obtained protein fraction was calculated as follows: Purity (%) = (100 %/(band intensity)all) * (band intensityABF).
Characterization
The samples obtained from each process step were analyzed by dynamic light scattering (DLS) to determine the particle sizes (Zetasizer NS, Malvern at 173° backscatter measurement with three repeated runs). Likewise, the Zeta potential was measured with the Zetasizer NS, Malvern at 293 K using laser Doppler anemometry for each sample. Samples from each process step were dried under vacuum conditions. The dried samples were measured by X-ray photoelectron spectroscopy (XPS, Karatos Axis Ultra DLD system). Elemental analysis was carried out with the dried powders, from the particles as received and after functionalization with GNTA on a FlashEA 1112 (ThermoQuest Italia S.p.A.). Transmission electron microscopy (TEM) measurements were carried out with a Zeiss EM9120 microscope operated at 120 kV using 200 square mesh carbon-coated copper grids (Plano). The condensation product of GNTA was characterized by attenuated total reflectance Fourier Transform infrared spectroscopy (ATR-FTIR) with a BRUKER EQUINOX 55. Raman spectroscopy was performed on a LabRam-HR 800 from Horiba at a wavelength of 532 nm using a nitrogen-cooled Spectrum One CCD-3000VCCD from Horiba.
Results and discussion
Characterization of surface-modified Fe2O3 nanoparticles
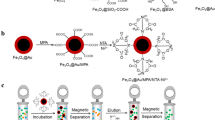
The magnetic nanoparticles were prepared by solvothermal synthesis using TEG as solvent as described formerly (Grabs et al. 2012). Subsequently, the condensation product GNTA was introduced onto the surface of the particles. A schematic illustration of the functionalization steps of the nanoparticles is shown in Fig. 1.

Schematic illustration of the preparation steps for the Ni2+-loaded functionalized nanoparticles
The plain nanoparticles, as obtained from the synthesis, were analyzed by Raman spectroscopy (Fig. 2). The spectrum confirmed that the core consisted mainly of iron oxide in the maghemite phase. To avoid the transformation of magnetite into hematite while measuring due to the heating of the sample by the laser source, a lens for light filtering was used. This resulted in a shift of the peaks to smaller wavelengths; nonetheless the sample seemed to consist rather of maghemite then magnetite (De Faria et al. 1997).

Raman spectrum of the obtained nanoparticles
Figure 3 shows TEM images of the samples after each process step. The TEM image of the magnetic nanoparticles dispersed in deionized water directly after the synthesis showed that the particles were rather spherical in shape with an average size of 8 nm (Fig. 3a). Due to the van der Waals forces, the particles tended to agglomerate. The aqueous particle samples were further modified by coupling variable amounts of the reaction product of GLYMO and NTA to the surface of the particles. As demonstrated in the TEM image (Fig. 3b) of the particles functionalized with 0.6 g GNTA, the particles are sterically stabilized, with the individual particles being well separated even after drying on the TEM grid. The addition of NiSO4 to the sample, obtaining Ni2+-charged particles, triggered agglomeration and large agglomerates with a size of several hundred nanometers were observed (Fig. 3c).

TEM image of the magnetic Fe2O3 nanoparticles a not functionalized in deionized water, b functionalized with GNTA, c functionalized with GNTA-Ni2+
Measurement of the Zeta potential revealed a potential of +22 mV for the unmodified particles in water. After binding GNTA onto the particle surface, the potential was −20 mV. The charging of the particles with Ni2+ ions changed the Zeta potential to −6 mV. This confirms the assumption that after addition of NiSO4 the particles agglomerate due to the decreased degree of repulsion between the particles.
The magnetic nanoparticle dispersions were characterized using DLS. The particle size distributions for the plain particles, the surface-modified Fe2O3 particles and the particles charged with Ni2+ ions, respectively, are shown in Fig. 4. The average hydrodynamic size based on the volume distribution x V,50 of the Fe2O3 nanoparticles was 18.2 nm; for the Fe2O3–GNTA-functionalized particles, the average size of the particles decreases to 10.1 nm. After charging the nanoparticles with Ni2+, the average size of the agglomerates increased to 1,500 nm. From the particle size distribution data, it can be concluded that washing and redispersion in water results in slight agglomeration of the particles. When covalently binding GNTA to the surface, the particles are sterically stabilized and the agglomerates are dissolved, resulting in a hydrodynamic size measured by DLS that essentially reflects the primary size. When adding the nickel salt, agglomerates were formed (>1 μm), as the zeta potential decreased with an increasing ion concentration. For comparison, a commercial system for magnetic purification by Promega® was analyzed. In contrast, these particles had a median size of x V,50 = 3,500 nm. Hence, the DLS results are in agreement with the TEM observations. To evaluate the binding of GNTA to the particle surface and to confirm the coupling between the silane and the ligand, high-resolution XPS of carbon, nitrogen, oxygen, silicon, and iron was carried out. The analysis of the Fe2p peak, as seen in Fig. 5a, confirmed that the magnetic nanoparticles consist of γFe 2 O 3. The Fe2p spectra show the typical structure for iron oxides with a broad, main, double-peak (Fe2p3/2 and Fe2p1/2), and typical shakeup satellites (McIntyre and Zetaruk 1977). The Fe2p3/2 peak was deconvoluted into two spectral bands at 711.14 and 713.76 eV. The peak shape and the lack of the low-binding energy shoulder observed for Fe 3 O 4 points to the sole presence of Fe(III) (Khurshid et al. 2009; Grosvenor et al. 1984; Minati et al. 2011). The most intense peak at 711.14 eV is attributed to iron in the maghemite phase; the peak at 713.76 eV represents hydroxides which can form on the surface of the particles due to reaction with moisture in the air. The Fe2p1/2 peak at 725.04 eV is attributed to the carboxylate–Fe bond. The binding of GNTA to the surface of the particles can be analyzed by looking at the O1s region in Fig. 5b and the Si2p region in Fig. 5c. For this purpose, the oxygen 1s peak was deconvoluted into three spectral bands at 529.47, 531.16, and 533.03 eV. Whereas the peak at 529.47 eV is attributed to Fe2O3, the encapsulation of the particles with GNTA led to a decrease of the intensity seen for the peak at 529.47 eV and an increase of the relative intensity of the peak at 533.03 eV, which can be attributed to the Si–O–Si groups. The peak at 531.16 eV can be attributed to O–H groups (Ki-Chul et al. 2006). The silicon 2p peak was deconvoluted into four spectral bands. The peaks located at the binding energies of 103.83, 102.70, 101.84, and 100.63 eV are characteristic for Si with different states of oxidation. The peak at 103.83 eV could be attributed to Si–C bonds while the other signals are typical for Si–O bonds (Suzer et al. 2010). The coupling between the silane and the ligand NTA can be analyzed by looking at the N2p region shown in Fig. 5d. The deconvolution of the N2p peak indicates the presence of secondary and tertiary amino groups at 399.14 and 400.49 eV, respectively (Durdureanu-Angheluta et al. 2012).

DLS analysis of Fe2O3 nanoparticles as obtained from the synthesis (dotted line), after functionalizing with Fe2O3–GNTA (dashed line) and after charging with Ni2+ ions (dashed and dotted line) and commercial particles (Promega®) (continuous line), all in deionized water

XPS spectra of the nanoparticles a Fe2p peak, b O1s peak, c Si2p peak, d N2p peak
The coverage of the iron oxide nanoparticle surface with GNTA was determined quantitatively with elemental analysis for nitrogen. The surface coverage was calculated on the basis of the surface area of the plain particles assuming a uniform particle size of 8 nm and spherical shape. For an amount of 0.6 g GNTA added to 100 mg nanoparticles, it was calculated that each particle is covered with about 2,000 GNTA molecules.
Ni-DAD analysis was conducted to determine gravimetrically the concentration of Ni2+ ions bound to the surface. The adsorption of Ni2+ ions onto the Fe2O3–GNTA particles was monitored as a function of added NiSO4. It was discovered that the amount of Ni2+ adsorbed to the Fe2O3–GNTA nanoparticles did not increase when increasing the concentration of NiSO4 (see S.I., Fig. 0.2). This suggested that the binding capacity of Fe2O3–GNTA had reached saturation when adding one molar equivalent NiSO4:NTA.
The Ni2+-charged magnetic nanoparticles were used to isolate the His-tagged ABF from the culture supernatant to evaluate their functionality for selective protein purification. First, the binding capacity of the Fe2O3–GNTA-Ni2+ toward 6-His-tagged proteins was evaluated using purified recombinant 6-His-tagged ABF D1.3 scFv. To obtain the binding capacity of the particles, increasing concentrations of ABF, ranging from 25 mgABF/L to 200 mgABF/L, were added to \(0.5\,{\text{g}}_{{\text{Fe}}_{2}{\text{O}}_{3}}/{\text{L}}\) magnetic nanoparticles each. The amount of ABF that was not bound to the particles was determined by SDS-PAGE and densitometric analysis by comparison to an ABF standard with a given concentration.
As shown in Fig. 6, the protein binding efficiency gradually increased with increasing concentration of ABF in suspension. For the highest dosage of 200 mgABF/L, 50 % of the product could not be isolated and remained in the supernatant. In contrast, a slightly lower binding capacity for the commercial Promega® system was observed, isolating only 45 % of the desired protein. Obviously, the usage of individual nanoparticles as developed here was superior to embedded ones as found in the Promega® system probably due to the high surface/volume ratio and a higher binding capacity. As a second step, the optimal protein separation performance of the particles was investigated by modulation of the added GNTA amount, varying from 0.5 to 3.0 g/100 mg Fe2O3. In a typical protein purification procedure, the functionalized particles were added to a pretreated cultivation supernatant, containing naturally secreted proteins and the recombinant ABF D1.3 scFv, to obtain a concentration of 0.24 mg/mL. Figure 7 shows the SDS-PAGE analysis of loaded particles from the two process steps, i.e., before (−) and after washing (+). Figure 7a reveals a defined band corresponding to the molecular weight of ABF D1.3 scFv before (−) and after washing (+) of the particles with almost the same intensity. This confirms that the particles were able to bind and selectively purify the His-tagged ABFs. Moreover, the target protein was not detached during washing, while the unspecific binding of other proteins was reduced. Figure 7b, c provides the densitometric analysis of the SDS-PAGE analysis. The concentration of GNTA, used to functionalize the particles, had a distinct effect on the amount of particle bound ABF and also on the purity of the separated protein. By alteration of the added amount of GNTA, the purity of the target protein ABF D1.3 scFv and its total amount varied. When increasing the amount of GNTA to 2.0 g, the relative amount of ABF bound decreased, whereas a further increase up to 3.0 g resulted in a somewhat higher amount of bound ABF. This may be attributed to several mechanisms: naturally, the increase of GNTA content would produce more docking sites available for binding to the particles. On the other hand, this could also result in a steric hindrance between the ligand and the antibody fragment, decreasing the amount of purified ABF. Moreover, the addition of NiSO4 in an equimolar ratio to NTA might trigger a stronger agglomeration, due to the higher ionic strength, hence decreasing the surface area available for purification. The detailed elucidation of the underlying mechanisms will be subject to our future investigations.

Adsorption isotherm for the binding of purified ABF D1.3 scFv to GNTA-Ni2+-functionalized magnetic nanoparticles (GLYMO + NTA-Ni2+ 0.6 g/Fe2O3 (100 mg))

Selective purification of ABF D1.3 scFv from a bacterial cultivation supernatant. The GNTA fraction was varied from 0.5 to 3.0 g/Fe2O3 (100 mg) and proteins bound to the particles (0.24 mg/mL) were analyzed by SDS-PAGE. Relative amounts of proteins were determined by densitometric analysis of protein bands. The purity was calculated related to the amount of unspecifically bound proteins
Since the absolute amount of ABF as well as its purity are aimed to be maximized, 0.6 g GNTA/100 mg (Fe2O3) was identified as an ideal GNTA ratio. Concentrations of ≥2 g GNTA/100 mg (Fe2O3) showed also feasible purification performance. However, these relatively high concentrations show barely higher amounts of adsorbed protein. Thus, the lower GNTA ratio was preferred in order to keep costs as low as possible.
Conclusion
In summary, a highly specific protein purification system with high capacity was prepared by immobilization of GNTA onto nanoparticles synthesized via the non-aqueous sol–gel method and subsequent charging with Ni2+ ions. The system exhibited a high adsorption capacity for the specific protein ABF D1.3 scFv while showing low adsorption of other proteins. The presented system thus has great potential to be used as an alternative separation system for His-tagged proteins or peptides as it can easily be prepared and shows high promise toward scale-up. The handling of the separation system is easy and separation is achieved rapidly with low nonspecific adsorption.
References
Anspach FB (1994) Silica-based metal chelate affinity sorbents. I. Preparation and characterization of iminodiacetic acid affinity sorbents prepared via different immobilization techniques. J Chromatogr A 672(1–2):35–49. doi:10.1016/0021-9673(94)80592-X
Arias JL, Lopez-Viota M, Ruiz MA, Lopez-Viota J, Delgado AV (2007) Development of carbonyl iron/ethylcellulose core/shell nanoparticles for biomedical applications. Int J Pharm 339(12):237–245. doi:10.1016/j.ijpharm.2007.02.028
Bucak S, Jones DA, Laibinis PE, Hatton TA (2003) Protein separations using colloidal magnetic nanoparticles. Biotechnol Prog 19(2):477–484. doi:10.1021/bp0200853
Chang J, Kang K, Choi J, Jeong Y (2008) High efficiency protein separation with organosilane assembled silica coated magnetic nanoparticles. Superlattices Microstruct 44(4–5):442–448. doi:10.1016/j.spmi.2007.12.006
Cristancho CAM, David F, Franco-Lara E, Seidel-Morgenstern A (2013) Discontinuous and continuous purification of single-chain antibody fragments using immobilized metal ion affinity chromatography. J Biotechnol 163(2):233–242. doi:10.1016/j.jbiotec.2012.08.022
Csetneki I, Faix MK, Szilagyi A, Kovacs AL, Nemeth Z, Zrinyi M (2004) Preparation of magnetic polystyrene latex via the miniemulsion polymerization technique. J Polym Sci A Polym Chem 42(19):4802–4808. doi:10.1002/pola.20300
David F, Steinwand M, Hust M, Bohle K, Ross A, Dübel S, Franco-Lara E (2011) Antibody production in Bacillus megaterium: strategies and physiological implications of scaling from microtiter plates to industrial bioreactors. Biotechnol J 6(12):1516–1531. doi:10.1002/biot.201000417
De Faria DLA, Venancio Silva S, De Oliveira MT (1997) Raman microspectroscopy of some iron oxides and oxyhydroxides. J Raman Spectrosc 28(11):873–878. doi:10.1002/(SICI)1097-4555(199711)28:11<873::AID-JRS177>3.0.CO;2-B
Duguet E, Vasseur S, Mornet S, Devoisselle JM (2006) Magnetic nanoparticles and their applications in medicine. Nanomedicine 1(2):157–168. doi:10.2217/17435889.1.2.157
Durdureanu-Angheluta A, Dascalu A, Fifere A, Coroaba A, Pricop L, Chiriac H, Tura V, Pinteala M, Simionescu BC (2012) Progress in the synthesis and characterization of magnetite nanoparticles with amino groups on the surface. J Magn Magn Mater 324(9):1679–1689. doi:10.1016/j.jmmm.2011.11.062
Faraji M, Yamini Y, Rezaee M (2010) Magnetic nanoparticles: synthesis, stabilization, functionalization, characterization, and applications. J Iran Chem Soc 7(1):1–37. doi:10.1007/BF03245856
Frenzel A, Fröde D, Meyer T, Schirrmann T, Hust M (2012) Generating recombinant antibodies for research, diagnostics and therapy using phage display. Curr Biotechnol 1:31–41. doi:10.2174/2211550111201010033
Ghosh Chaudhuri R, Paria S (2011) Core/shell nanoparticles: classes, properties, synthesis mechanisms, characterization, and applications. Chem Rev 112(4):2373–2433. doi:10.1021/cr100449n
Grabs IM, Bradtmöller C, Menzel D, Garnweitner G. (2012) Formation mechanisms of iron oxide nanoparticles in different nonaqueous media. Cryst Growth Des 12(3):1469–1475. doi:10.1021/cg201563h
Grosvenor AP, Kobe BA, Biesinger MC, McIntyre NS (2004) Investigation of multiplet splitting of Fe2p XPS spectra and bonding in iron compounds. Surf Interface Anal 36(12):1564–1574. doi:10.1002/sia.1984
Gu H, Xu K, Xu C, Xu B (2006) Biofunctional magnetic nanoparticles for protein separation and pathogen detection. Chem Commun 9:941–949. doi:10.1039/B514130C
Gupta AK, Gupta M (2005) Synthesis and surface engineering of iron oxide nanoparticles for biomedical applications. Biomaterials 26(18):3995–4021. doi:10.1016/j.biomaterials.2004.10.012
Hainfeld JF, Liu W, Halsey CMR, Freimuth P, Powell RD (1999) Ni-NTA gold clusters target His-tagged proteins. J Struct Biol 127(2):185–198. doi:10.1006/jsbi.1999.4149
Heyd M, Franzreb M, Berensmeier S (2011) Continuous rhamnolipid production with integrated product removal by foam fractionation and magnetic separation of immobilized Pseudomonas aeruginosa. Biotechnol Prog 27(3):706–716. doi:10.1002/btpr.607
Käppler T, Cerff M, Ottow K, Hobley T, Posten C (2009) In situ magnetic separation for extracellular protein production. Biotechnol Bioeng 102(2):535–545. doi:10.1002/bit.22064
Khurshid H, Kim SH, Bonder MJ, Colak L, Ali B, Shah SI, Kiick KL, Hadjipanayis GC (2009) Development of heparin-coated magnetic nanoparticles for targeted drug delivery applications. J Appl Phys 105(7):07B308. doi:10.1063/1.3068018
Ki-Chul K, Eung-Kwon K, Jae-One L, Young-Sung K (2006) Characterization of magnetic nanoparticles synthesized by sonomechanical method. In: Nanotechnology Materials and Devices Conference, 2006. NMDC 2006. IEEE, vol 1, pp 600–601. doi:10.1109/NMDC.2006.4388922
Lee IS, Lee N, Park J, Kim BH, Yi YW, Kim T, Kim TK, Lee IH, Paik SR, Hyeon T (2006) Ni/NiO core/shell nanoparticles for selective binding and magnetic separation of histidine-tagged proteins. J Am Chem Soc 128(33):10,658–10,659. doi:10.1021/ja063177n
Lee S, Ahn C, Lee J, Lee J, Chang J (2012) Rapid and selective separation for mixed proteins with thiol functionalized magnetic nanoparticles. Nanoscale Res Lett 7:279. doi:10.1186/1556-276X-7-279
Low D, O’Leary R, Pujar NS (2007) Future of antibody purification. J Chromatogr B 848(1):48–63. doi:10.1016/j.jchromb.2006.10.033
McIntyre NS, Zetaruk DG (1977) X-ray photoelectron spectroscopic studies of iron oxides. Anal Chem 49(11):1521–1529. doi:10.1002/sia.1984
Minati L, Micheli V, Rossi B, Migliaresi C, Dalbosco L, Bao G, Hou S, Speranza G (2011) Application of factor analysis to XPS valence band of superparamagnetic iron oxide nanoparticles. Appl Surf Sci 257(24):10,863–10,868. doi:10.1016/j.apsusc.2011.07.123
Mornet S, Vasseur S, Grasset F, Duguet E (2004) Magnetic nanoparticle design for medical diagnosis and therapy. J Mater Chem 14(14):2161–2175. doi:10.1039/b402025a
Palecek E, Fojta M (2007) Magnetic beads as versatile tools for electrochemical DNA and protein biosensing. Talanta 74(3):276–290. doi:10.1016/j.talanta.2007.08.020
Roque A, Cecilia A, Lowe CR, Taipa MA (2004) Antibodies and genetically engineered related molecules: production and purification. Biotechnol Prog 20(3):639–654. doi:10.1021/bp030070k
Schneider CA, Rasband WS, Eliceiri KW (2012) NIH image to ImageJ: 25 years of image analysis. Nat Methods 9(7):671–675. doi:10.1038/nmeth.2089
Shieh DB, Su CH, Chang FY, Wu YN, Su WC, Hwu J, Chen JH, Yeh CS (2006) Aqueous nickel-nitrilotriacetate modified Fe3O4–NH +3 nanoparticles for protein purification and cell targeting. Nanotechnology 17(16):4174–4182. doi:10.1088/0957-4484/17/16/030
Sopaci S, Simsek I, Tural B, Volkan M, Demir A (2009) Carboligation reactions with benzaldehyde lyase immobilized on superparamagnetic solid support. Org Biomol Chem 7(8):1658–1664. doi:10.1039/b819722a
Suzer S, Baer DR, Engelhard MH (2010) Analysis of Fe nanoparticles using XPS measurements under d.c. or pulsed-voltage bias. Surf Interface Anal 42(6–7):859–862. doi:10.1002/sia.3260
Xu C, Xu K, Gu H, Zheng R, Liu H, Zhang X, Guo Z, Xu B (2004) Dopamine as a robust anchor to immobilize functional molecules on the iron oxide shell of magnetic nanoparticles. J Am Chem Soc 126(32):9938–9939. doi:10.1021/ja0464802
Acknowledgments
The authors thank Prof. G. Goya and G. Antorrena Pardo, University of Zaragoza, Spain, for the XPS analysis, and D. Menzel, Institute for Condensed Matter Physics, TU Braunschweig, for the Raman analysis. R. Pitschke and Prof. M. Antonietti, Max Planck Institute for Colloids and Interfaces, Potsdam, are gratefully acknowledged for the TEM measurements. We also thank Miriam Steinwand and Prof. Stefan Dübel, Institute of Biochemistry, Biotechnology and Bioinformatics, for provision of the D1.3 scFv antibody standard. This work was kindly supported by the German Research Foundation via the Collaborative Research Center (SFB 578-From Gene to Product) at TU Braunschweig.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Masthoff, IC., David, F., Wittmann, C. et al. Functionalization of magnetic nanoparticles with high-binding capacity for affinity separation of therapeutic proteins. J Nanopart Res 16, 2164 (2014). https://doi.org/10.1007/s11051-013-2164-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-013-2164-6