
Abstract
Chitosan was conjugated with folic acid (FA) and the resulting chitosan derivatives with a FA-substitution degree of around 6 % was used to synthesize FA-conjugated chitosan–polylactide (FA–CH–PLA) copolymers to build a drug carrier with active targeting characteristics for the anticancer drug of paclitaxel (PTX). Selected FA–CH–PLAs with various polylactide percentages of about 40 wt% or lower were employed to fabricate nanoparticles using sodium tripolyphosphate as a crosslinker, and different types of nanoparticles were endued with similar average particle-sizes located in a range between 100 and 200 nm. Certain types of PTX-loaded FA–CH–PLA nanoparticles having encapsulation efficiency of around 90 % and initial load of about 12 % were able to release PTX in a controlled manner with significant regulation by polylactide content in FA–CH–PLAs. Targeting characteristic of achieved nanoparticles was confirmed using FA-receptor-expressed MCF-7 breast cancer cells. The uptake of PTX revealed that optimized FA–CH–PLA nanoparticles with an equivalent PTX-dose of around 1 μg/mL could have more than sixfold increasing abilities to facilitate intracellular paclitaxel accumulation in MCF-7 cells after 24 h treatment as compared to free PTX. At a relatively safe equivalent PTX-dose for normal MCF-10A mammary epithelial cells, the obtained results from Hoechst 33342 staining indicated that optimized PTX-loaded FA–CH–PLA nanoparticles had more than threefold increasing abilities to induce MCF-7 cell apoptosis in comparison to free PTX.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Paclitaxel (PTX), a natural antitumor drug usually extracted from the bark of the Pacific yews, has been widely used in the clinical treatments for a variety of tumors such as oophoroma, metastatic breast cancer, and non-small cell lung cancer (Spratlin and Sawyer 2007). PTX is currently formulated with Cremophor EL (polyoxyl 35 castor oil) and ethanol to increase its solubility for clinical applications because PTX is poorly soluble in aqueous medium (Javeed et al. 2009). However, utilization of diluents and solubilizer for PTX during intravenous administration gives rise to several problems (Kim et al. 2006; Marupudi et al. 2007): (1) easily occurred precipitation of PTX due to solvent changeover; (2) certain side effects of hypersensitivity reactions arisen from the use of Cremophor EL, including neurotoxicity, nephrotoxicity, cardiac toxicity, and so on; and (3) possibly released plasticizers extracted from intravenous infusion lines due to the use of organic solvents. In view of these problems, alternative carriers for intravenous formulation of PTX need to be developed in order to circumvent the use of Cremophor EL and ethanol while effectively increasing the bioavailability of PTX.
Up to now, various systems for delivery of PTX, including liposomes, cyclodextrins, emulsions, mixed micelles, microspheres, and polymeric nanoparticles, have been investigated to avoid side effects of Cremophor EL (Kim et al. 2006; Marupudi et al. 2007; Greco and Vicent 2009). In point of vehicle materials for delivery of drugs, biocompatible and biodegradable polymers obtained from synthetic or natural sources have attracted a great deal of attention due to their tailorable structures and properties as well as processability in favor of various vehicle forms (Greco and Vicent 2009; Locatelli and Franchini 2012). Of naturally occurring polymers being abundant in nature, chitosan has been considered to be an approving material to serve as drug vehicles because it has several advantageous features, including biocompatibility, biodegradability, hydrophilicity, non-toxicity, nonantigenicity, bioadherence, and cell affinity as well (Sinha et al. 2004; Wan and Wen 2005). In the case of antitumor drug delivery, chitosan and its derivatives have already been utilized as carriers in the form of nanomicells (Liu et al. 2010a; Yao et al. 2007), gels (Bhattarai et al. 2010), or nanoparticles (Duan et al. 2012; Santos et al. 2011). Nevertheless, in consideration of high hydrophobicity of PTX, chitosan alone seems not to be competent enough for delivering PTX with required initial loading in well-controlled manner even though some doable crosslinkers have already been employed because chitosan is quite hydrophilic. Some studies have thus directed toward modification of chitosan to achieve necessitated derivatives with certain amphiphilic properties. To date, chitosan-based carriers for PTX delivery have mainly been modified at the C-2 site of chitosan backbone using certain hydrophobic segments and less attention has been paid to hydroxyl groups at the C-6 or C-3 site of chitosan units (Pan et al. 2013; Zhang et al. 2003, 2009). In addition, these PTX-loaded chitosan derivative carriers are usually used in a passive-targeting manner.
Grafting hydrophobic chains onto the C-6 site of chitosan backbone and leaving the amino groups at the C-2 site free may generate some desired copolymers that can act as an amphiphilic carrier for loading hydrophobic drugs while being further modified at the C-2 site of chitosan units with preferred groups or molecules for active-targeting delivery of drugs.
Folic acid receptor is known to be abundantly expressed on the surface of different human epithelial cancer cells, including cancers of the ovary, kidney, uterus, colon, and lung, whereas in most normal tissue it is expressed in very low levels (Low and Antony 2004; Elnakat and Ratnam 2004). Conjugation of drugs or active macromolecules with folic acid can thus enhance drug endocytosis or even develop targeting ability toward certain cancer cells. In fact, folic acid has already functioned as a promising ligand for some vehicles that are used for delivering antitumor drugs specifically toward to folate-receptor expressed cancer cells (Morris and Sharma 2010; Li et al. 2010). In the case of chitosan-based drug carriers, folate-conjugated chitosan nanoparticles have been also utilized as potential vehicles for targeting HT-29 cells (Li et al. 2011) as well as for targeted delivery of 5-aminolaevulinic acid (Yang et al. 2010). In addition, folate-modified carboxymethyl chitosan nanoparticles have also been examined for targeted delivery of doxorubicin (Sahu et al. 2010).
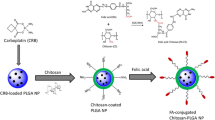
Despite the rapidly growing variety of vehicles for delivering different drugs, nano-scale active-targeting carriers with satisfactory performance for intravenous administration of PTX are still very few. In the present study, an attempt was made to synthesize a type of folate-conjugated chitosan–polylactide amphiphilic copolymers, and then, to fabricate them into PTX-loaded nanoparticles in an aqueous medium system. It is expected that the hydrophobic core-like interior of ternary nanoparticles can function as a favorable portion for loading PTX, whereas the folate-conjugates on the surface of nanoparticles will facilitate folate-mediated intracellular uptake of PTX toward folate-receptor expressed cancer cells. Thus, efficiently intravenous administration of PTX could benefit from our carriers. Some results related to synthesis, characterization, and in vitro examination of newly developed nanoparticle carriers were reported.
Materials and methods
Materials
Chitosan powder [degree of deacetylation: 91.4 %; viscosity average MW: 6.1 × 105, measured following reported methods (Wan et al. 2004)] was supplied by Aladdin Inc. l-lactide (LA) from Sigma-Aldrich was recrystallized from dry toluene and vacuum-dried prior to use. Paclitaxel was purchased from Tocris Bioscience. N-hydroxy succinimide (NHS), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), folate (folic acid, FA), Hoechst 33342, dicyclohexyl carbodiimide (DCC), and some biochemical reagents were purchased from Sigma-Aldrich. All other chemicals were of analytical grade and purchased from different commercial sources.
Synthesis of folate-conjugated chitosan (FA–CH)
FA–CHs were synthesized via a two-step pathway. Active NHS ester of FA was first prepared following reported methods with some modifications (Guo et al. 1999; Zhu et al. 2011). Briefly, 0.68 m mol of LA was dissolved in 10 mL anhydrous DMSO, 0.1 mL of triethylamine, equimolar NHS and DCC were added. The reaction was allowed to proceed overnight at 30 °C under stirring and shielded from light. The byproduct dicyclohexylurea was removed by filtration. The mixture was concentrated to some extent using a rotary evaporator and the product was obtained by precipitation using cold acetone (40 %) in ether. The precipitates were repeatedly washed with ether and dried in vacuum.
In a typical procedure for synthesis of FA–CHs, 100 mg of chitosan was dissolved in 20 mL of acetic acid/sodium acetate buffer (pH 4.7), and to this solution, 5 mL DMSO solution of NHS-folate (20 mg/mL) were added with stirring at 40 °C in the dark for 12 h. The reaction system was brought to pH 9.0 by dropwise addition of diluted aqueous NaOH to precipitate the mixture. The product was extensively washed with distilled water, re-dissolved in 2 % aqueous acetic acid solution, and the unreacted folic acid in the product was removed using a Sephadex G-25 column (UV detector: 363 nm; elution solvent: 2 % aqueous acetic acid solution; flow-rate: 1.5 mL/min). Different FA–CHs were synthesized by varying the feed ratio of NHS-folate to chitosan.
Synthesis of folate-conjugated chitosan–polylactide (FA–CH–PLA)
The FA–CH with a FA-substitution degree of around 6 wt% was used for synthesis of FA–CH–PLAs. Polylactide (PLA) was grafted onto the FA–CH following group-protection methods with some modifications (Duan et al. 2010; Zhou et al. 2013; Feng and Dong 2007). In a typical procedure, 1 g of FA–CH was dissolved in 30 mL of methanesulfonic acid, followed by addition of 2.57 g of LA monomer and 0.73 g of 4-dimethylaminopyridine. The reaction was allowed to perform for 12 h at 120 °C with stirring while bubbling with nitrogen. The resulting mixture was then transferred to a beaker that contained 200 mL of 0.2 M KH2PO4, 20 mL of 4 M NaOH and was cooled in a bath (0 °C) to remove the acid residue. The collected precipitate was extracted with toluene to remove PLA homopolymers. Afterward, the product was dialyzed against distilled water for 2 days using a membrane tube (MW cutoff: 10kD) and lyophilized for further use. By mainly changing the feed ratio of LA to FA–CHs, several types of FA–CH–PLA copolymers were synthesized using the same protocol. In addition, some chitosan–polylactide (CH–PLA) copolymers without FA-conjugation were also synthesized with the same technique and they were used to prepare CH–PLA nanoparticles.
Preparation of nanoparticles
Selected FA–CH–PLAs with PLA content of around 40 wt% or lower were used for fabricate PTX-loaded nanoparticles and a typical example preparation procedure was as follows: 30 mg of FA–CH–PLA was dissolved in 12 mL of acetic acid/sodium acetate buffer, and to this solution, 2 mL of 0.2 % PTX ethanol solution was added and well mixed with stirring. Afterward, the mixture was slowly added with 6 mL of 0.1 % sodium tripolyphosphate (TPP) solution using a 21 G needle with stirring at 30 °C for 1 h. The resulting nanoparticle suspension was centrifuged at 22,000 rpm for 30 min, and the retrieved product was repeatedly washed with distilled water until the pH became around 7.0. The collected nanoparticles were then lyophilized at −80 °C for further measurements. Different types of FA–CH–PLA nanoparticles were prepared by mainly changing the PLA percentage in FA–CH–PLAs and the volume of acetic acid/sodium acetate buffer, while maintaining the feed ratio of TPP to matrix at a fixed rate of 1:4, in order to enable the resulting nanoparticles to have similar average particle-sizes.
Some PTX-loaded chitosan, PTX-loaded FA–CH, blank CH–PLA, and blank FA–CH–PLA nanoparticles were also prepared using the same method. In addition, certain types of nanoparticles were labeled with FITC under light protection by adding FITC methanol solution during the preparation of nanoparticles following the same protocol mentioned above, and they were used for certain examinations related to cell uptake.
Characterization
Infrared (IR) spectra of different powder samples were recorded on a spectrophotometer (VERTEX 70).
A serial of FA solutions with stepwise-changed volumes varying from 0.5 to 3.0 mL (step-size: 0.5 mL) was taken from a 0.01 wt% FA solution in acetate buffer, and each was diluted up to 10 mL. Diluents were assayed at 363 nm using a UV spectrophotometer to generate a fitted straight line that functions as a standard. Afterward, a given amount of FA–CH samples was dissolved in the same acetate buffer and FA-substitution degrees for different FA–CHs were determined according to the standard.
FA–CH or FA–CH–PLA samples were dissolved in a mixed solvent of deuterated water and deuterated trifluoroacetic acid (97:3, v/v) to produce solutions with the same concentration (Liu et al. 2010b), and their 1H NMR spectra were measured on an NMR spectrometer (Bruker Avance 600).
The weight percentage of PLA in FA–CH–PLAs was measured using a gravimetric method described elsewhere (Feng and Dong 2006).
Different types of nanoparticles were suspended in deionized water to produce very dilute solutions, and the solutions were sonicated to well disperse the nanoparticles. A drop of sample solution was placed onto 300 mesh carbon-coated copper grid. After 10 min of deposition, the grid was tapped with blotting paper, stained with 2 % uranyl acetate and dried before loading for TEM examination.
Colloidal nanoparticle suspensions were prepared using ultrapure water and they were evaluated by dynamic light scattering measurements using an instrument (Zetasizer Nano ZS) to determine hydrodynamic particle sizes. The zeta potential (ζ) of nanoparticles was also measured on the same instrument.
Determination of encapsulation efficiency and loading efficiency
Encapsulation efficiency (EE) and loading efficiency (LE) for different types of nanoparticles were determined with a spectrophotometric method. 5 mg of PTX-loaded nanoparticles was dispersed in ethanol and vortexed for 12 h at 37 °C. The supernatant was collected by centrifugation at 20,000 rpm for 30 min and assayed at 227 nm. EE and LE were calculated as follows:
where M o is the mass of PTX encapsulated inside nanoparticles, M 1 refers to the mass of totally added PTX, and M denotes the mass of nanoparticles.
In vitro release of paclitaxel
PTX was dissolved in ethanol and diluted with a mixture of ethanol and distilled water containing 0.5 % Tween 80 to prepare a series of solutions with varied concentrations increasing from 1 × 10−6 to 20 × 10−6 mol/L with a step-size of 5 × 10−6 mol/L. These solutions were measured at 227 nm to establish a concentration-absorbance standard for the quantification of released PTX.
A given amount of PTX-loaded nanoparticles was dispersed in 2 mL PBS solution (pH 7.4), and the resulting bulk solution was introduced into a dialysis tube (MW Cutoff: 5000). The sealed dialysis tube was immerged in 50 mL PBS buffer (pH 7.4) containing 0.5 % Tween 80 and 10 % ethanol with stirring at 60 rpm and 37 °C. At prescribed intervals, 5 mL of release medium was withdrawn and an isometric amount of fresh medium was refilled. PTX in the release medium was extracted using 5 mL of methylene dichloride. After fully volatilizing the solvent, the released PTX was dissolved in 1 mL of ethanol for HPLC analysis (Kromasil C18 column: 250 × 4.6 mm, 5 μm; mobile phase: a mixture of methanol, acetonitrile and ultra pure water; flow rate: 1.0 mL/min; detection wavelength: 227 nm; 30 °C).
Cell viability
Human breast cancer cells (MCF-7) were cultured in FA-free RPMI1640 medium supplement with 10 % fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37 °C in an atmosphere of 5 % CO2 with relative humidity at 95 %. Normal human mammary epithelial cell line (MCF-10A) was grown in medium MEGM (mammary epithelial growth medium, serum-free) supplemented with cholera toxin in a humidified atmosphere containing 5 % CO2 at 37 °C. Cultured medium for each type of cells was replaced on alternate days until confluence. After that, cells were trypsinized and collected by centrifugation at 1,000 rpm for 5 min. The harvested cells were resuspended in their respective media for further experiments.
The cell viability was assessed using a MTT assay. Aliquots of MCF-7 and MCF-10A cells were seeded into 96-well culture plates (1 × 104 cells/well) with their own media and incubated for 24 h. Afterward, each well was refreshed with culture medium, respectively, containing equivalent dose of free PTX and PTX-loaded nanoparticles at various amounts changing from 1 to 10,000 ng/mL. Blank CH–PLA nanoparticles and blank FA–CH–PLA nanoparticles were also tested as vehicle controls. After 48-h incubation, 15 μL of MTT (5 mg/mL in PBS) solution was added to each well and additionally incubated for 4 h at 37 °C. The media was then aspirated and 150 μL of DMSO was added to dissolve the formazan crystals. The absorbance of each well was read at 570 nm using a microplate reader.
Cell uptake
MCF-7 cells were seeded into two groups of 24-well culture plates (2 × 105 cells/well) and preincubated with FA-free RPMI1640 medium for 24 h. Cells in group one were then exposed to free FITC or FITC-labeled blank nanoparticles at a final equivalent FITC concentration of 2.0 μg/mL and cultured for 2–4 h at 37 °C. To see the effect of free folate competition on the cell uptake, some wells were added with 2 mM FA. After incubation for the matched period of time, cells in each well were washed three times with PBS before imaging with a fluorescence microscope.
In the case of group two, cells were cultured with equivalent dose of free PTX and PTX-loaded nanoparticles for various periods up to 24 h. At predetermined time points, the medium was removed, and cells were trypsinized and lysed with ethanol using probe sonication so that PTX internalized into cells was well extracted. The resulting lysate was separated by centrifugation at 12,000 rpm for 10 min, and the supernatant containing PTX was quantified using HPLC analysis under the same running conditions as mentioned for PTX release measurements. Percent uptake of PTX was determined by the ratio of the internalized PTX to the fed equivalent dose of PTX.
Apoptosis analysis of cells
Cell apoptosis was assessed using fluorescent DNA-binding dye Hoechst 33342. Cells (MCF-7 or MCF-10A, 2 × 105/well) were seeded onto sterile cover slips placed in 6-well culture plates containing respective media. After 24-h incubation, cells were treated with equivalent dose (PTX concentration: 20 ng/mL) of free PTX and PTX-loaded nanoparticles, and further cultured for various periods up to 72 h. Cells were then fixed in a methanol/acetic acid (3:1) mixture at 4 °C for 10 min. Fixed cells were stained with 10 μg/mL Hoechst 33342 in the dark for 20 min. Cells were washed with PBS three times and viewed under a fluorescence microscopy. To quantify the apoptosis of the cells, at least 500 cells were counted per experiment, and percent apoptosis was defined as follows:
Normal nuclei were identified as non-condensed chromatin while apoptotic nuclei were identified as condensed chromatin or fragmented nuclei.
Statistical analysis
Data are presented as mean ± standard deviation. In the case of normal population, means were compared by Student’s t tests, and otherwise, one-way analysis of variance with post-hoc multi-comparison methods was performed with p < 0.05 as the criteria for statistical significance.
Results and discussion
Synthesis of folate-conjugated chitosan–polylactide
In the present study, FA–CH–PLAs were synthesized by first conjugating FA onto chitosan and then the resulting FA–CH was grafted with PLA side chains. In addition, PLA side chains were grafted onto the C-6 sites of chitosan backbone rather than the C-2 sites using methanesulfonic acid as a group protective reagent (Duan et al. 2010) in considering that surplus amino groups at the C-2 site of FA–CHs can be effectively crosslinked by TPP based on preparatory trials.
Figure 1 presents several representative IR spectra for chitosan, FA–CH, and FA–CH–PLA. A band at 1,596 cm−1 and a shoulder at round 1,655 cm−1 in Fig. 1a are typical bands for chitosan having a relatively high deacetylation degree and they can be respectively attributed the N–H bending of primary amine and C=O stretch of secondary amine of chitosan (Wan et al. 2008a; Wu et al. 2011a). In the case of spectrum for FA–CH, a band at 1,585 cm−1 is ascribed to the residual primary amine of chitosan backbone with a small shift in comparison with chitosan; a new peak appeared at 1,633 cm−1 can be considered as a shift of the band for secondary amine originally registered at round 1,655 cm−1 due to the overlap of newly formed C–N bond during amidation between FA and chitosan (Li et al. 2011); two other peaks at 1,460 and 1,425 cm−1 may be ascribed to the presence of aromatic ring in FA. These results confirm that FA has been conjugated onto chitosan. With respect to FA–CH–PLA (see Fig. 1c), new peaks appear at 1,752 and 1,241 cm−1, and they should be assigned to the ester or carboxylic groups in branched PLA chains (Duan et al. 2010; Liu et al. 2010b; Yao et al. 2003). The peak located at around 1,582 cm−1 in Fig. 1c indicates that the residual amino groups at the C-2 site of FA–CHs still remain, revealing that PLA chains are grafted onto C-6 sites of FA–CHs instead of C-2 sites.

Representative IR spectra of chitosan a, FA–CH b and FA–CH–PLA c (FA-substitution degree: 9.3 % for FA–CH and FA–CH–PLA; PLA percentage in FA–CH–PLA: 31.9 wt%)
Figure 2 shows typical 1H NMR spectra for chitosan, FA–CH, and FA–CH–PLA samples. In Fig. 2a, chemical shifts at around 2.58 (H-2), 3.2–3.6 (H-3, -4, -5, -6), and 4.8 (H-1) ppm can be attributed to the chitosan (Liu et al. 2010b). With respect to FA–CH, δ signals at about 1.5, 2.3, 3.0–3.4, and 4.8 ppm are assigned to the resonance of the monosaccharide residue protons, respectively (Li et al. 2011; Yang et al. 2010; Dube et al. 2002); and several signals appearing at 6.51–8.42 ppm (aromatic ring) and the singlet at 4.19 ppm can be attributed to folate (Dube et al. 2002; Wang et al. 1997). In Fig. 2c, the new peak at about 1.2 ppm can be ascribed to the methyl protons located at the terminal groups and the backbone of the polylactide moiety, whereas another new singlet at ca. 4.1 ppm can be assigned to the protons in the repeat units of PLA chains (Liu et al. 2010b; Wu et al. 2011b).

1H NMR spectra of chitosan a, FA–CH b and FA–CH–PLA c (FA-substitution degree: 9.3 % for FA–CH and FA–CH–PLA; PLA percentage in FA–CH–PLA: 39.1 wt%)
In comparison to the spectrum of chitosan, FA–CH–PLA has the characteristic peaks for chitosan backbone with small shifts in its spectrum, and meanwhile, the peak area ratio of H-2 to H-1 remains almost the same, suggesting that PLA side chains are grafted onto the C-6 site of FA–CH main chains. Based on results of IR and 1H NMR measurements, it can be reached that FA–CH and FA–CH–PLA with designed structures have been successfully synthesized.
In the present study, FA was conjugated onto chitosan using active NHS ester of FA rather than direct use of FA itself, and a maximum FA-substitution degree of about 17.1 wt% for some FA–CHs was achieved under optimal reaction conditions. However, some reports revealed that certain protein molecules showed strong enough targeting characteristics toward different FA-receptor-expressed cells when each of them carried only three FA-conjugates (Low and Antony 2004; Lu and Low 2002). In our case, the FA-substitution degree for FA–CHs would be around 1.4 wt% if a chitosan molecule chain is conjugated with three folate molecules. In considering a fact that certain amounts of FA-conjugates on FA–CH–PLAs could be trapped inside FA–CH–PLA nanoparticles and they would possibly lose their targeting functions, a FA-substitution degree of 1.4 wt% seems not to be high enough to play a targeting role for the resulting nanoparticles. Some FA–CHs with a FA-substitution degree of 6.1 ± 0.17 wt% were thus selected as starting materials for the synthesis of FA–CH–PLAs on the grounds of many trials.
Under present synthetic conditions, PLA content in FA–CH–PLAs can reach around 47 wt% or even higher. Nevertheless, PLA content needs adjusting within a proper range to effectively control amphiphilic properties of FA–CH–PLAs because otherwise FA–CH–PLAs containing a higher PLA percentage will not be well soluble in aqueous solvents and may also result in poor fabrication of nanoparticles. FA–CH–PLAs with PLA content of about 40 wt% or lower were therefore chosen for fabricating PTX-loaded FA–CH–PLA nanoparticles based on certain tentative experiments.
Folate-conjugated chitosan–polylactide nanoparticles
Different types of covalent crosslinkers, such as diisocyanate, epoxy compound, and glutaraldehyde have been commonly used to crosslink chitosan so far (Liao et al. 2013; Wu et al. 2011b). In view of many free amino groups in FA–CH–PLAs, these crosslinkers are also applicable for crosslinking FA–CH–PLAs. Nevertheless, in the present case, TPP, a type of ionic crosslinker, was employed in consideration of clinic safety of TPP because the mentioned covalent crosslinkers for chitosan can impair biocompatibility of the resulting FA–CH–PLA nanoparticles (Bao et al. 2008; Liao et al. 2013). Figure 3a shows a representative TEM image for some FA–CH–PLA nanoparticles and the size-distribution of nanoparticles is presented in Fig. 3b. It can be reached that nanoparticles showed regular sphericity without aggregation and their size was distributed between several tens of nanometers and around 280 nm with an approximate Gaussian-distribution character. Despite complex processing parameters, several variables, such as PLA content in FA–CH–PLAs, concentration of FA–CH–PLA solutions, fed amount of TPP and stirring speed, were found to be crucial for controlling the sizes and properties of FA–CH–PLA nanoparticles. These variables were thus optimized using orthogonal experimental design to produce required nanoparticles having similar average particle-sizes located in a range between 100 and 200 nm, and relevant parameters for resulting nanoparticles are summarized in Table 1.

TEM image a and size-distribution b of FA–CH–PLA nanoparticles (FA-substitution degree: 6.1 %; PLA content in FA–CH–PLA: 30.1 wt%; and the feed ratio of TPP to matrix: 1/4)
Table 1 shows that different types of nanoparticles had their average particle-size changing from around 129–142 nm with a good acceptable polydispersity ranging between 0.237 and 0.346. Data in Table 1 exhibit that all types of nanoparticles had positive zeta potential varying from around 21–27 mV, suggesting that these nanoparticles will have practical stability in a colloidal system since the absolute value of zeta potential is higher than 15 mV (Zhong et al. 2012). The positive zeta potential for these nanoparticles should be ascribed to the contribution of positive charge arisen from residual amino groups in chitosan units. In comparison to NP-I, NP-II nanoparticles had lower zeta potential (p < 0.05), which can be attributed a fact that FA–CH was used for building NP-II nanoparticles and it had less amino groups as compared to chitosan since some amino groups that are related to positive zeta potential were already consumed in virtue of FA-conjugates. With respect to samples in set NP-III, there were no significant differences in their zeta potential. The samples in set NP-III were produced using FA–CH–PLAs with various PLA contents but FA-substitution degree and feed ratio of TPP to matrix were retained the same. Insignificant changes in zeta potential for three types of samples in set NP-III may be due to similar structure formed during the nanoparticle preparation. FA–CH–PLAs are amphiphilic polymers, and hence, hydrophilic chitosan chains could stretch outward and shape as hydrophilic outer layer, while PLA side chains would be entangled together as close as possible to form the hydrophobic interior regardless of PLA content in FA–CH–PLAs because these nanoparticles were prepared in aqueous media (Wan et al. 2008b). As a result, positive charges arisen from surplus amino groups exposed on the surface of FA–CH–PLA nanoparticles would contribute to positive zeta potential and the PLA content in FA–CH–PLAs would have insignificant impacts on the zeta potential of nanoparticles. As shown in Table 1, FA–CH–PLA nanoparticles had increasing EE and LE as the PLA content in FA–CH–PLAs increased, which can be ascribed to a fact that higher PLA content in FA–CH–PLAs would generate a higher ratio of hydrophobic interior volume to the integral volume of nanoparticles, leading to increasing EE and LE for hydrophobic PTX. It worthy of mentioning that LE of some types of FA–CH–PLA nanoparticles reached as high as around 12 %, suggesting that they have high potential to function as carriers with practical significance of application.
In vitro release of paclitaxel
To see PTX release behavior of nanoparticles, samples in set NP-III were examined while NP-I, NP-II, and NP-IV nanoparticles were used as controls. Figure 4 illustrates release profiles for relevant samples. Plots for NP-I and NP-II indicate that these nanoparticles released the loaded PTX at very fast release rates and cumulative amount of released PTX was higher than 95 % during 2 days release. In consideration of very low EE and LE and uncontrollable release behavior of NP-I and NP-II nanoparticles, they were excluded from further investigations because they are evidently unsuitable for delivering PTX. Samples in set NP-III had greatly reduced initial burst in the first day, depending on the PLA content in FA–CH–PLAs. After one-day release, each type of nanoparticles in set NP-III released PTX in a sustained manner at similar release rates for a few days, and their release behavior was significantly regulated by the PLA content in FA–CH–PLAs. In view of relatively rigid and hydrophobic characteristics of PLA side chains (Wan and Wen 2005; Wan et al. 2008b), PLA in FA–CH–PLAs may function as hook-like branches inside nanoparticles to drag the entrapped PTX molecules and hinder them from diffusing cross the surface of nanoparticles, resulting in reduced initial burst and subsequently delayed release. Higher PLA content in FA–CH–PLAs will endow the resulting FA–CH–PLA nanoparticles with increasing abilities to slow down the PTX release.

Paclitaxel release patterns for different types of nanoparticles (see Table 1 for parameters of nanoparticles)
Figure 4 shows that the release profiles of NP-IV nanoparticles was almost the same as that of NP-III(3) nanoparticles. These results may be due to the high similarity between them in their respective sizes and properties. As indicated in Table 1, a main difference between NP-IV and NP-III(3) nanoparticles was that the former was prepared with CH–PLA and the latter was produced using FA–CH–PLA containing around 6 wt% of FA-conjugates. Nevertheless, both NP-IV and NP-III(3) nanoparticles had very similar parameters such as mean-size, PDI, EE, and LE with an insignificant difference in their zeta potential. PLA content in both CH–PLA and FA–CH–PLA was about 40 wt% while FA-conjugates in FA–CH–PLA was only around 6 %, the release profiles of NP-IV and NP-III(3) nanoparticles could be predominantly controlled by the PLA content, and thus, FA-conjugates had insignificant impacts on them as a result.
In the light of higher EE and LE as well as well-controlled release behavior of NP-III(3) and NP-IV nanoparticles, some PTX-loaded NP-III(3) and NP-IV nanoparticles, blank NP-III(3) and NP-IV nanoparticles and their corresponding FITC-labeled nanoparticles were thus selected for following cell experiments.
Cell uptake
It is known that free PTX shows various degrees of cytotoxicity to many different types of cells even at a relatively low dose, and FA-conjugated nanoparticles loaded with PTX may result in increasing complexity during the internalization of nanoparticles due to the effect arisen from both FA-conjugates and PTX itself. Some FITC-labeled blank nanoparticles (NP-V and NP-VI nanoparticles, see Table 1) were therefore used for measuring their cellular uptake while an equivalent amount of free FITC was used as control to find out whether FA-conjugates would effectively influence the cellular uptake of nanoparticles. In addition, a certain amount of free folate was used as a competition reagent for FA-conjugated nanoparticles to see if free folate regulates the uptake of nanoparticles.
Uptake measurements were conducted using FA-receptor over-expressed MCF-7 cells (Lee et al. 2005) and some representative fluorescence images for the cellular uptake versus culture time are presented in Fig. 5a. The uptake of free FITC was very low and did not significantly change during incubation with cell monolayers. Column-two in Fig. 5a shows that the uptake of NP-V nanoparticles without the built-in FA targeting component was enhanced in comparison to free FITC and it visually increased as the culture time went on. In the case of NP-VI nanoparticles with FA-conjugates, their uptake clearly increased as compared to the corresponding one for NP-V nanoparticles. In the presence of free folate competition (column-four in Fig. 5a), the uptake of NP-VI nanoparticles visibly diminished in comparison to that without folate competition competes (column-three in Fig. 5a) and it reached almost the same levels when comparing with the matched one in column-two. These results suggest that FA-conjugates in FA–CH–PLA nanoparticles can function as active-targeting ligands to significantly enhance intracellular uptake of FA–CH–PLA nanoparticles and the internalization of FA–CH–PLA nanoparticles into MCF-7 cells is regulated via a FA-receptor-mediated pathway.

Fluorescence images for uptake of free FITC and FITC-labeled blank nanoparticles internalized in MCF-7 cells (a, incubation time: 2–4 h; original magnification: 400×; see Table 1 for matched parameters); and percent PTX uptake in MCF-7 cells incubated with free PTX and PTX-loaded nanoparticles (b, incubation time: 24 h)
Assessment of PTX accumulation in MCF-7 cells was performed using free PTX, PTX-loaded NP-IV and NP-III(3) nanoparticles and relevant data are depicted in Fig. 5b. In comparison to free PTX, NP-IV nanoparticles had enhanced ability to transport PTX into MCF-7 cells, and on the other hand, the PTX uptake delivered by NP-III(3) nanoparticles showed a more than sixfold increase when the equivalent dose of PTX reached 1,000 ng/mL or higher. It has been reported that folic acid can be transported into different cells via potocytosis, a receptor-mediated endocytosis, involving a series of steps (Lee 2006). In this process, the ligand-bound receptor is first sequestered in caveolae, internalized into postcaveolar plasma vesicles, released from the receptor via an intravesicular reduction in pH, and subsequently transported into the cytoplasm. After that, the ligand-free receptor will be recycled back to the cell surface by reopening of the caveolae (Hilgenbrink and Low 2005). Figure 5a confirms that blank FA–CH–PLA nanoparticles had well-defined targeting characteristics, and hence, the high PTX uptake for NP-III(3) nanoparticles can be ascribed to the FA-receptor-mediated interactions between the FA-receptors on the surface of MCF-7 cells and NP-III(3) nanoparticles. Although NP-IV nanoparticles were prepared using CH–PLA without FA-conjugates (see Table 1), the surface of NP-IV nanoparticles carries positive charge and is hydrophilic, which may facilitate the internalization of NP-IV nanoparticles via a passive entrapping pathway, resulting in relatively high PTX uptake by using NP-IV nanoparticles as a carrier when compared to free PTX.
Cell viability
Blank nanoparticles were first tested to see their effect on the viability of MCF-10A and MCF-7 cells against the nontoxic controls and relevant results are represented in Fig. 6a. The bar-graphs in Fig. 6a exhibit that after being treated with blank nanoparticles for 48 h, more than 90 % of cells remained viable within the measurement range, confirming that blank NP-III(3) or NP-IV nanoparticles in an amount up to 100 μg/mL are nearly non-toxic to MCF-10A or MCF-7 cells.

Viability of MCF-10A and MCF-7 cells (48-h incubation). a Treatment with blank NP-IV and NP-III(3) nanoparticles; and b treatment with free PTX, and PTX-loaded NP-IV and NP-III(3) nanoparticles
The effect of free PTX and PTX-loaded nanoparticles on the viability of MCF-10A and MCF-7 cells is presented in Fig. 6b. It can be seen that MCF10A cells with minimal FA-receptor expression (Lee 2006) and MCF-7 cells with FA-receptor over-expression were not significantly affected by the treatment of free PTX and PTX-loaded NP-IV or NP-III(3) nanoparticles when the equivalent PTX dose was 1 ng/mL, and as the equivalent dose increased, free PTX and PTX-loaded NP-IV or NP-III(3) nanoparticles imposed different impacts on the viability of MCF-10A or MCF-7 cells.
The viability of MCF-10A cells decreased from around 75, 86, and 85 % to about 9, 28, and 25 %, respectively corresponding to free PTX, NP-IV, and NP-III(3) nanoparticles when the equivalent PTX dose increased from 10 to 10,000 ng/mL, revealing that free PTX had significantly stronger cytotoxicity (p < 0.01) against MCF-10A cells as compared to NP-IV or NP-III(3) nanoparticles loaded with the same equivalent PTX dose. In the case of MCF-10A cells, FA-receptor-mediated internalization of PTX-loaded NP-IV or NP-III(3) nanoparticles can be negligible because of the minimal FA-receptor-expression in MCF-10A cells. In comparison to free PTX, the much lower cytotoxicity of PTX-loaded NP-IV or NP-III(3) nanoparticles against MCF-10A cells could be attributed the delayed release of PTX entrapped inside nanoparticles. As shown in Fig. 4, both NP-IV and NP-III(3) nanoparticles released only about 30 % of loaded PTX during 48 h in vitro incubation, respectively, meaning that in the present instance, the PTX amount accumulated from intracellular and/or extracellular release from NP-IV or NP-III(3) nanoparticles would be lower than the same equivalent amount of free PTX. As a result, NP-IV or NP-III(3) nanoparticles showed relatively low cytotoxicity to MCF-10A cells as compared to free PTX.
In the case of MCF-7 cells, PTX-loaded NP-III(3) nanoparticles exhibited significantly increasing ability to inhibit the growth of MCF-7 cells in comparison to free PTX or NP-IV nanoparticles. The cytotoxicity of NP-III(3) nanoparticles reached more than twofold or even threefold higher in MCF-7 cells at an equivalent PTX dose of 1,000 or 10,000 ng/mL, as compared to free PTX. Despite the delayed release of PTX from NP-III(3) nanoparticles, the uptake of PTX in MCF-7 cells was more than sixfold higher as compared to free PTX during 24-h incubation when NP-III(3) nanoparticles were used as vehicle and the equivalent PTX dose was 1,000 ng/mL or higher (see Fig. 5b). The synergistical effect stemmed from NP-III(3) nanoparticles might be much stronger than that of free PTX at the same equivalent PTX dose, and hence, NP-III(3) nanoparticles resulted in significantly higher cytotoxicity to MCF-7 cells. With respect to NP-IV nanoparticles, they showed a relatively low ability to inhibit the growth of MCF-7 cells when comparing to free PTX at the equivalent PTX dose of around 10 ng/ML or higher, which may be caused by two factors. One is related to a fact that the uptake of PTX transported by NP-IV nanoparticles was only double that of free PTX in the same PTX-dose range (see Fig. 5b), and another one is associated with delayed PTX release from NP-IV nanoparticles (see Fig. 4). The superimposed results of two factors could lead to a lower growth-inhibiting ability of NP-IV nanoparticles against MCF-7 cells.
Above presented results indicate that NP-III(3) nanoparticles are able to well retain the bioactivity of entrapped PTX and selectively deliver PTX to the targeted cells. At a relatively safe equivalent PTX dose used for normal cells, for example, 100 ng/mL, the majority of MCF-10A cells with treatments remained viable, whereas the growth of MCF-7 cells with the same treatment was already greatly inhibited, suggesting that the presently developed FA–CH–PLA nanoparticles with optimized parameters have promising potential for delivering hydrophobic PTX specifically toward FA-receptor over-expressed cells while meeting certain safety requirements.
Induction of apoptosis in cells
PTX is able to induce cell death involving in many types of cells via apoptosis pathway which is mainly related to a microtubule disruption mechanism (Sudo et al. 2004). To see the effect of nanoparticles on cell apoptosis, MCF-7 and MCF-10A cells were exposed to an equivalent PTX dose of 20 ng/mL for various periods of time, respectively, and stained with Hoechst 33342 using unexposed cells as controls. Figure 7a shows that the control MCF-10A cells had a regular oval-shaped nucleus body and their nuclei were homogeneously stained with dim appearance. Upon exposure to free PTX or NP-III(3) nanoparticles for 48 h, some MCF-10A cells exhibited typical apoptotic features such as nucleus deformation, nucleus shrinkage, and chromatin condensation with enhanced fluorescence (Cao et al. 2007). Like the control MCF-10A cells, the control MCF-7 cells were in the shape of regular oval and stained dimly, while the MCF-7 cells treated with free PTX or NP-III(3) nanoparticles were intensely stained and exhibited morphologic changes in their nucleus patterns, including nucleus shrinkage, chromatin condensation, and even nucleus fragmentation. In addition, it can also be observed that in the case of MCF-7 cells, NP-III(3) nanoparticles had an increasing ability to induce the cell apoptosis as compared to free PTX.

Apoptosis profiles of MCF-10A and MCF-7 cells exposed to free PTX and PTX-loaded nanoparticles at an equivalent PTX dose of 20 ng/mL. a Nucleus patterns of cells stained by Hoechst 33342 (a control MCF-10A cells without treatment of PTX, b MCF-10A cells + free PTX, c MCF-10A cells + NP-III(3) nanoparticles, d control MCF-7 cells without treatment of PTX, e MCF-7 cells + free PTX, and f: MCF-7 cells + NP-III(3) nanoparticles, incubation time: 48 h, original magnification: 200×); and b percent apoptosis of cells versus incubation time (asterisk indicates a significant difference, p < 0.05)
Figure 7b presents quantitative measurements for percent apoptosis of cells. After treatment with free-PTX or NP-III(3) nanoparticles at an equivalent PTX dose of 20 ng/mL for various durations up to 72 h, free PTX had similar effect on the apoptosis rate of MCF-10A or MCF-7 cells at different measurement points (p > 0.05). On the other hand, NP-III(3) nanoparticles imposed significantly different impacts on the apoptosis rate of MCF-10A or MCF-7 cells. In comparison to free PTX, NP-III(3) nanoparticles induced significantly less apoptosis in MCF-10A cells after 48 h treatment or longer (p < 0.05), whereas they showed considerably increasing ability to induce MCF-7 cell apoptosis. In particular, after 72-h incubation, around 85 % of MCF-10A cells treated with NP-III(3) nanoparticles were still viable while the percent apoptosis of NP-III(3) nanoparticle-treated MCF-7 cells reached higher than 70 %, indicating that more than four times of MCF-7 cells became apoptotic as compared to MCF-10A cells. In addition, it can also be seen that the number of apoptotic MCF-7 cells treated with NP-III(3) nanoparticles for 72 h was more than threefold higher than that treated with free PTX. These results suggest that within a relative safe equivalent PTX dose range for normal MCF-10A mammary epithelial cells, the PTX-loaded FA–CH–PLA nanoparticles have a remarkably enhanced ability to induce apoptosis of MCF-7 breast cancer cells in comparison to free PTX.
Conclusions
FA–CH–PLAs with a FA-substitution degree of around 6 % and various PLA percentages of around 40 wt% or lower were successfully synthesized. The resulting FA–CH–PLA nanoparticles were suited for loading hydrophobic paclitaxel with high EE and initial paclitaxel loading. Some paclitaxel-loaded FA–CH–PLA nanoparticles had certain abilities to control the release of paclitaxel, and PLA content in FA–CH–PLAs was able to significantly regulate release rates of nanoparticles. FA-conjugates in FA–CH–PLAs was effective to facilitate internalization of FA–CH–PLA nanoparticles into FA-receptor-expressed MCF-7 breast cancer cells through a FA-receptor-mediated pathway, and optimized FA–CH–PLA nanoparticles were capable of significantly enhancing intracellular paclitaxel accumulation in MCF-7 cells as compared to free paclitaxel. The entrapped paclitaxel inside the nanoparticles well retained its bioactivity, and at a relatively safe equivalent PTX dose for normal MCF-10A mammary epithelial cells, certain PTX-loaded FA–CH–PLAL nanoparticles had more than threefold increasing abilities to induce the apoptosis of MCF-7 breast cancer cells in comparison to free PTX, suggesting that presently developed FA–CH–PLA nanoparticle has a promising potential for targeted delivery of hydrophobic PTX.
References
Bao H, Li L, Zhang H (2008) Influence of cetyltrimethylammonium bromide on physicochemical properties and microstructures of chitosan-TPP nanoparticles in aqueous solutions. J Colloid Interface Sci 328:270–277
Bhattarai N, Gunn J, Zhang M (2010) Chitosan-based hydrogels for controlled, localized drug delivery. Adv Drug Delivery Rev 62:83–99
Cao J, Liu Y, Jia L, Zhou HM, Kong Y, Yang G, Jiang LP, Li QJ, Zhong LF (2007) Curcumin induces apoptosis through mitochondrial hyperpolarization and mtDNA damage in human hepatoma G2 cells. Free Radical Biol Med 43:968–975
Duan K, Chen H, Huang J, Yu J, Liu S, Wang D, Li Y (2010) One-step synthesis of amino-reserved chitosan-graft-polycaproctone as a promising substance of biomaterial. Carbohydr Polym 80:498–503
Duan J, Liu M, Zhang Y, Zhao J, Pan Y, Yang X (2012) Folate-decorated chitosan/doxorubicin poly(butyl)cyanoacrylate nanoparticles for tumor-targeted drug delivery. J Nanopart Res 14:761–769
Dube D, Francis M, Leroux JC, Winnik FM (2002) Preparation and tumor cell uptake of poly(N-isopropylacrylamide) folate conjugates. Bioconjug Chem 13:6856–6892
Elnakat H, Ratnam M (2004) Distribution, functionality and gene regulation of folate receptor isoforms: implications in targeted therapy. Adv Drug Delivery Rev 56:1067–1084
Feng H, Dong CM (2006) Preparation and characterization of chitosan-graft- poly(ε-caprolactone) with an organic catalyst. J Polym Sci Part A 44:5353–5361
Feng H, Dong CM (2007) Synthesis and characterization of phthaloyl-chitosan -g-poly(L-lactide) using an organic catalyst. Carbohydr Polym 70:258–264
Greco F, Vicent MJ (2009) Combination therapy: opportunities and challenges for polymer-drug conjugates as anticancer nanomedicines. Adv Drug Deli Rev 61:1203–1213
Guo W, Hinkle GH, Lee RJ (1999) 99mTcHyNICFolate: a novel receptor-based targeted radiopharmaceutical for tumor imaging. J Nuclear Med 40:1563–1569
Hilgenbrink AR, Low PS (2005) Folate receptor-mediated drug targeting: from therapeutics to diagnostics. J Pharm Sci 94:2135–2146
Javeed A, Ashraf M, Riaz A, Ghafoor A, Afzal S, Mukhtar MM (2009) Paclitaxel and immune system. Euro J Pharm Sci 38:283–290
Kim J, Kim Y, Kim S, Park JH, Kim K, Choi K, Chung H, Jeong SY, Park R, Kim I, Kwon IC (2006) Hydrophobically modified glycol chitosan nanoparticles as carriers for paclitaxel. J Control Release 111:228–234
Lee YK (2006) Preparation and characterization of folic acid linked poly(L-glutamate) nanoparticles for cancer targeting. Macromol Res 14:387–393
Lee ES, Na K, Bae YH (2005) Doxorubicin loaded pH-sensitive polymeric micelles for reversal of resistant MCF-7 tumor. J Control Release 103:405–418
Li N, Yang X, Zhai G, Li L (2010) Multifunctional pluronic/poly(ethylenimine) nanoparticles for anticancer drug. J Colloid Interface Sci 350:117–125
Li P, Wang Y, Zeng F, Chen L, Peng Z, Kong LX (2011) Synthesis and characterization of folate conjugated chitosan and cellular uptake of its nanoparticles in HT-29 cells. Carbohydr Res 346:801–806
Liao C, Huang J, Sun S, Xiao B, Zhou N, Yin D, Wan Y (2013) Multi-channel chitosan-polycaprolactone conduits embedded with microspheres for controlled release of nerve growth factor. React Funct Polym 73:149–159
Liu J, Li H, Jiang X, Zhang C, Ping Q (2010a) Novel pH-sensitive chitosan-derived micelles loaded with paclitaxel. Carbohydr Polym 82:432–439
Liu L, Shi A, Guo S, Fang Y, Chen S, Li J (2010b) Preparation of chitosan-g-polylactide graft copolymers via self-catalysis of phthaloylchitosan and their complexation with DNA. React Funct Polym 70:301–305
Locatelli E, Franchini MC (2012) Biodegradable PLGA-b-PEG polymeric nanoparticles: synthesis, properties, and nanomedical applications as drug delivery system. J Nanopart Res 14:1316–1334
Low PS, Antony AC (2004) Folate receptor-targeted drugs for cancer and inflammatory diseases. Adv Drug Delivery Rev 56:1055–1058
Lu Y, Low PS (2002) Folate-mediated delivery of macromolecular anticancer therapeutic agents. Adv Drug Delivery Rev 54:675–693
Marupudi NI, Han JE, Li KW, Renard VM, Tyler BM, Brem H (2007) Paclitaxel: a review of adverse toxicities and novel delivery strategies. Expert Opin Drug Safety 6:609–621
Morris VB, Sharma CP (2010) Folate mediated in vitro targeting of depolymerised trimethylated chitosan having arginine functionality. J Colloid Interface Sci 348:360–368
Pan Z, Gao Y, Heng L, Liu Y, Yao G, Wang Y, Liu Y (2013) Amphiphilic N-(2, 3-dihydroxypropyl)- chitosan-cholic acid micelles for paclitaxel delivery. Carbohydr Polym 94:394–399
Sahu SK, Mallick SK, Santra S, Maiti TK, Ghosh SK, Pramanik P (2010) In vitro evaluation of folic acid modified carboxymethyl chitosan nanoparticles loaded with doxorubicin for targeted delivery. J Mater Sci Mater Med 21:1587–1597
Santos DP, Ruiz MA, Gallardo V, Zanoni MVB, Arias JL (2011) Multifunctional antitumor magnetite/chitosan-l-glutamic acid (core/shell) nanocomposites. J Nanopart Res 13:4311–4323
Sinha VR, Singla AK, Wadhawan S, Kaushik R, Kumria R, Bansal K, Dhawan S (2004) Chitosan microspheres as a potential carrier for drugs. Int J Pharm 274:1–33
Spratlin J, Sawyer MB (2007) Pharmacogenetics of paclitaxel metabolism. Critical Rev Oncol Hematol 61:222–229
Sudo T, Nitta M, Saya H, Ueno NT (2004) Dependence of paclitaxel sensitivity on a functional spindle assembly checkpoint. Cancer Res 64:2502–2508
Wan Y, Wen D (2005) Preparation and characterization of porous conducting poly(DL-lactide) composite membranes. J Membr Sci 246:193–201
Wan Y, Creber KAM, Peppley B, Bui VT (2004) Structure and ionic conductivity of a series of di-o-butyrylchitosan membranes. J Appl Polym Sci 94:2309–2323
Wan Y, Cao X, Wu Q, Zhang S, Wang S (2008a) Preparation and mechanical properties of poly(chitosan-g-DL-lactic acid) fibrous mesh scaffolds. Polym Adv Technol 19:114–123
Wan Y, Cao X, Zhang S, Wang S, Wu Q (2008b) Fibrous poly(chitosan-g-DL-lactic acid) scaffolds prepared via electro-wet-spinning. Acta Biomater 4:876–886
Wang S, Luo J, Lantrip DA, Waters DJ, Mathias CJ, Green MA, Fuchs PL, Low PS (1997) Design and synthesis of [111In] DTPA-folate for use as a tumor-targeted radiopharmaceutical. Bioconjug Chem 8:673–679
Wu H, Zhang J, Xiao B, Zan X, Gao J, Wan Y (2011a) N-(2-hydroxypropyl)-3-trimethylammonium chitosan-poly(ε-caprolactone) copolymers and their antibacterial activity. Carbohydr Polym 83:824–830
Wu J, Liao C, Wang Z, Cheng W, Zhou N, Wang S, Wan Y (2011b) Chitosan-polycaprolactone microspheres as carriers for delivering glial cell line-derived neurotrophic factor. React Funct Polym 71:925–932
Yang SJ, Lin FH, Tsai KC, Wei MF, Tsai HM, Wong JM, Shieh MJ (2010) Folic acid-conjugated chitosan nanoparticles enhanced protoporphyrin IX accumulation in colorectal cancer cells. Bioconjug Chem 21:679–689
Yao F, Chen W, Wang H, Liu H, Yao K, Sun P, Lin HA (2003) Study on cytocompatible poly(chitosan-g-L-lactic acid). Polymer 44:6435–6441
Yao Z, Zhang C, Ping QN, Yu L (2007) A series of novel chitosan derivatives: synthesis, characterization and micellar solubilization of paclitaxel. Carbohydr Polym 68:781–792
Zhang C, Ping QN, Zhang HJ, Shen J (2003) Preparation of N-alkyl-O-sulfate chitosan derivatives and micellar solubilization of taxol. Carbohydr Polym 54:137–141
Zhang Y, Huo M, Zhou J, Yu D, Wu Y (2009) Potential of amphiphilically modified low molecular weight chitosan as a novel carrier for hydrophobic anticancer drug: synthesis, characterization, micellization and cytotoxicity evaluation. Carbohydr Polym 77:231–238
Zhong L, Fu S, Peng X, Zhan H, Sun R (2012) Colloidal stability of negatively charged cellulose nanocrystalline in aqueous systems. Carbohydr Polym 90:644–649
Zhou N, Zan X, Wang Z, Wu H, Yin D, Liao C, Wan Y (2013) Galactosylated chitosan-polycaprolactone nanoparticles for hepatocyte-targeted delivery of curcumin. Carbohydr Polym 94:420–429
Zhu J, Zhou Z, Yang C, Kong D, Wan Y, Wang Z (2011) Folate-conjugated amphiphilic star-shaped block copolymers as targeted nanocarriers. J Biomed Mater Res Part A 97A:498–508
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No. 81071470).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Huang, S., Wan, Y., Wang, Z. et al. Folate-conjugated chitosan–polylactide nanoparticles for enhanced intracellular uptake of anticancer drug. J Nanopart Res 15, 2096 (2013). https://doi.org/10.1007/s11051-013-2096-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-013-2096-1