
Abstract
Periodontal disease affects tooth-supporting structures and nanoparticles (NPs) have been a promising approach for its treatment. The purpose of the study was to develop triclosan-loaded poly-ε-caprolactone (PCL) NPs for the treatment of periodontal infections. Solvent displacement method was used to prepare NPs following Box–Behnken design. The NPs were evaluated with respect to particle size, polydispersity index, surface morphology, zeta potential, thermal properties, in vitro drug release, and cell viability assay. The optimized NPs were in the size range of 180–230 nm with a mean size of 205.61 ± 10.4 nm. Entrapment efficiency (EE) of 91.02 ± 2.4 % was obtained with a drug loading of 21.71 ± 1.3 %. About 97 % of drug was released in vitro after 3 h. NPs demonstrated almost 100 % cell viability in L929 cell lines. Shelf life of the nanoparticles was 17.07 months. PCL affected particle size whereas triclosan affected loading and EE. The optimized NPs were spherical with smooth surface and exhibited biphasic in vitro release pattern. NPs had optimum zeta potential and PDI and were stable on storage. Absence of cytotoxicity of NPs to L929 cells indicated its safety. Triclosan-loaded PCL nanoparticles could thus serve as a novel colloidal drug delivery system against periodontal infections.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Periodontal disease is a localized pathological condition affecting the tooth-supporting structures which include the gingiva, periodontal ligament, cementum, and alveolar bone (Southard and Godowski 1998; Schwach-Abdellaoui et al. 2000; Pihlstrom et al. 2005). The main area involved in periodontal disease is the gingival sulcus, which is a pocket between the teeth and the gums and is called as periodontal pocket (Schroeder 1976). This pocket occurs as a result of localized Gram-negative anaerobic pathogenic bacterial infection below the gum line. If left untreated, these conditions results in the destruction of the periodontal ligament, resorption of the alveolar bone and the migration of the junctional epithelium along the tooth surface. This in turn, leads to loosening and subsequent loss of teeth (Ozmeric 2004). However, host defense mechanisms have been reported to play a role in the destruction process, which is triggered by numerous inflammatory and immune mediators of bacterial and host origin (Jain et al. 2008).
The modern periodontal therapy involves the removal of bacterial deposits (plaque and calculus) from the tooth surface (Schwach-Abdellaoui et al. 2000; Ozmeric 2004). This is accomplished by mechanical scaling and root planning. But this procedure alone does not always eliminate the pathogenic bacteria due to their presence within periodontal tissues, or in case of deeper pockets, their inaccessibility to the instrumentation (Slots and Rams 1990). Therefore, systemic or local treatment with antimicrobial agents has to be supplemented. However, systemic antibiotic therapy, especially over a long period of time raises the risk of undesirable side effects such as nausea, diarrhea, fever, abdominal pain, pseudomembranous colitis (Southard and Godowski 1998), hypersensitivity, and the development of bacterial resistance (Bollen and Quirynen 1996). These obvious drawbacks made the use of systemic antibiotic therapy increasingly unpopular.
To date, a significant number of approaches for the management of periodontal disease have been proposed. But unfortunately, the difficulty in accessing the periodontal pocket due to the complexity in anatomy of the root and the contours of the lesion, which leads to poor penetration of junctional epithelium by some of these systems has rendered most of it only partially successful (Medlicott et al. 1994; Schwach-Abdellaoui et al. 2000; Piñón-Segundo et al. 2005). This uncertainty of the conventional delivery system on periodontal disease treatment has significantly increased the interest of researchers to focus on nanotechnology-based approaches, i.e., colloidal drug carriers, and it has proved to be promising. Among these carriers, NPs, niosomes and liposomes have been most extensively investigated.
The advantages of NPs which includes smaller size, ability of site specificity, stability during storage as compared with liposomes, improved dissolution and bioavailability makes it a suitable delivery system for the treatment of periodontal disease. In addition, studies on the transport of NPs through the junctional epithelium have revealed that NPs can provide a potential intrapocket carrier system for the delivery of active substances to the periodontal pocket (Piñón-Segundo et al. 2005).
In the present work, NPs were selected as a drug delivery system due to its ability to penetrate junctional epithelium and gingival sulcus (main area involved in periodontal disease), and also as it has been proved promising in providing and maintaining an effective drug release rate in the periodontal pocket. Triclosan was selected as the drug to be incorporated into the NPs due to the following reasons: Triclosan is a broad spectrum antimicrobial agent that has a recognized efficacy against several plaque-forming bacteria, the main causative of periodontal infections (Rosling et al. 1997; Chiappetta et al. 2008). PCL was selected as the polymer to prepare NPs for the treatment of periodontal infections as it possesses several advantages that include: biocompatibility, biodegradability, high encapsulation capacity, non-toxicity, absence of generation of an acidic environment during degradation as compared to polylactides and glycolides and its slow degradation as compared to PLGA and polyglycolic acid-co-lactic acid, which makes it more suitable for controlled drug delivery (Koleske 1978; Murthy 1997; Sinha et al. 2004).
The solvent displacement method (Espuelas et al. 1997; Quintanar-Guerrero et al. 1998) was used to prepare NPs. Box–Behnken statistical design was used to optimize NPs containing triclosan. This design is suitable for exploring quadratic responses and constructing second-order polynomial models. The experimental design consists of a set of points lying at the midpoint of each edge and the replicated center point of the multidimensional cube that defines the region of interest. The independent and dependent variables are defined and listed in Table 1. The polynomial equation generated by this experimental design is as follows:
where R is the dependent variable, N 0 is the intercept, N 1–N 9 are the regression coefficients, and A, B, and C are the independent variables.
Materials and methods
Triclosan was procured from Ajay Organics Pvt. Ltd., Gujarat, India. Poly-ε-caprolactone (M w 14,000) was purchased from Sigma-Aldrich Co., MO, USA. Pluronic F-68 was purchased from Sigma-Aldrich Chemie, Munich, Germany. Brij® 35 (Polyoxyethylene lauryl ether) was obtained from Merck Specialities Pvt. Ltd., Mumbai, India. Mouse fibroblast cells of the permanent cell line L929 were procured from NCCS, Pune, India. Acetone was purchased from SD Fine-chemicals Limited, Mumbai, India. Box–Behnken statistical design with three factors, three levels, and 17 runs was used for the optimization of nanoparticles and Design-Expert® 8.0.0.6 (State-Ease Inc, Minneapolis, USA) software was used for statistical evaluation of the design. All other chemicals used in this study were of analytical grade and were used without further purification.
Preparation of nanoparticles
The NPs were prepared by solvent displacement method (Espuelas et al. 1997; Quintanar-Guerrero et al. 1998). All the batches were prepared according to the experimental design shown in Table 2. Accurately weighed amounts of PCL and triclosan were dissolved in acetone by mild heating and sonication. The polymer–drug mixture solution was gently injected drop wise into 30 ml of aqueous solution of Pluronic® F-68 under magnetic stirring. Stirring was continued until acetone was evaporated off completely. The resultant suspension of NPs was centrifuged at 13,000 rpm for 1 h. The supernatant was discarded after determining the entrapment efficiency (EE). The pellet was washed twice with distilled water and then freeze-dried using Heto Dry Winner Freeze dryer (Heto-Holten, Denmark).
Optimization
The optimized formula was determined after studying the effects of the independent variables on the responses. The criteria followed for generating the optimized formula were based on selecting the individual variable and defining it goal and limits. After defining the constraints for each variable, the Design Expert® software generated the optimized formula. NPs were prepared based on the optimized formula and then the evaluation studies were carried out.
Particle size, polydispersity index, and zeta potential
For each batch of nanoparticle suspension prepared, samples were analyzed in triplicate for particle size, polydispersity index, and zeta potential after suitable dilution with distilled water. Analysis was performed with a Nano ZS Zetasizer (Malvern Instruments Ltd., Worcestershire, United Kingdom) at a temperature of 25 °C. The instrument was equipped with software for particles size analysis, polydispersity index, and zeta potential. The measurements were carried out in triplicate.
Percentage drug loading (DL) and entrapment efficiency
A known weight of the lyophilized triclosan-loaded PCL NPs was dissolved in 2 mL acetone and then mixed with approximately 5 mL of 0.1 M NaOH. The organic solvent (acetone) was evaporated by stirring with magnetic stirrer. The resultant suspension was filtered and the volume was adjusted to 10 mL with 0.1 M NaOH. Triclosan content was assayed spectrophotometrically at 293 nm (UV-1601, SHIMADZU, MD, USA). Placebo NPs were prepared and processed in the same manner and were used as blanks. Determinations of DL and EE were done in triplicate.
DL and EE were calculated according to the following equations:
Scanning electron microscopy (SEM)
For SEM analysis of the freeze-dried NPs, the sample was mounted on an aluminum sample mount and sputter-coated with gold–palladium alloy to minimize surface charging. SEM analysis was performed using a LEO 435 V scanning electron microscope (Leo Electron Microscopy Ltd., Cambridge, UK) at a working distance of 15 mm and an accelerating voltage of 15 kV. The surface morphology of the particles was observed on different magnifications.
Transmission electron microscopy (TEM)
The morphology and size of the NPs were confirmed by TEM using Morgagni 268(D) TEM (FEI™ Company, Oregon, USA). In this method, a drop of the NPs suspension was placed on copper electron microscopy grids and then negative stained with phosphotungstic acid solution. After about 30 s, the sample was washed with ultra-purified water and the excess fluid was removed with a piece of filter paper. The dried sample was then examined and the resulting images were recorded.
Differential scanning calorimetry (DSC)
Differential scanning calorimetry analysis was carried out on triclosan, PCL and freeze-dried NPs samples. The test was carried out in order to define the physical state of the drug and the polymer in the NPs and to detect any drug–polymer interactions within the polymeric network of the NPs. In each case, the dried sample (about 2 mg) was loaded and sealed into aluminum pan with the help of DSC loading puncher. Each sample was scanned between 30 and 200 °C at a heating rate of 10 °C/min, under nitrogen atmosphere, using a differential scanning calorimeter (Pyris 6 DSC)—Pyris 6 DSC, Perkin Elmer, California, USA. The resultant thermograms were recorded.
In vitro release studies
As triclosan is almost insoluble in the aqueous medium with a solubility of <0.01 g/L at 25 °C, 2 % (w/v) Brij® 35 (Polyoxyethylene lauryl ether) was added in distilled water in order to perform in vitro release studies of triclosan. Besides increasing the solubility, the use of Brij® 35 aqueous solution as dissolution medium also prevented the adsorption of triclosan to the container surfaces (Chawla and Amiji 2002; Piñón-Segundo et al. 2005).
An accurate weight of lyophilized NPs was suspended in 500 mL of Brij® 35 aqueous solution using USP dissolution apparatus (VDA-8DR, VEEGO, Mumbai, India). The suspension was paddle-stirred at 50 rpm. The temperature was maintained at 37 ± 2 °C. At selected time intervals, 4 mL of the samples were withdrawn using a syringe adapted to a 0.10-μm syringe filter and replaced by fresh medium to maintain sink conditions. The absorbance of the samples was measured spectrophotometrically at 283 nm. The release experiments were carried out in triplicate.
Cell viability assay
Dubelco’s minimal essential medium (DMEM) supplemented with 10 % (v/v) fetal calf serum, Penicillin (500 IU) and streptomycin (250 mg mL−1) antibiotics was used for the study. Mouse fibroblast cells of the permanent cell line L929 (NCCS, Pune, India) were routinely propagated in DMEM containing 10 % fetal calf serum, Penicillin (100U mL−1), and streptomycin (100 μg mL−1) at 37 °C in an air atmosphere containing 5 % CO2 and then stored in cell culture medium containing 10 % dimethyl sulfoxide over liquid nitrogen until use. Repeated cytotoxicity experiments were done in identical stock cultures.
For cytotoxicity testing, 2.5 × 105 cells/well were seeded in 96-well ELISA plates in triplicates and incubated for 24 h at 37 °C in an air atmosphere containing 5 % CO2. After incubation the supernatants were removed and replaced with solutions/dispersions of free drug as well as control and optimized nanoparticulate formulations in DMEM media (200 μL) having equivalent concentration in triplicates to the 96-well plates. Then the cells were incubated for 24 h. Thereafter, the medium were removed and 100 μL of MTT solution (0.5 mg mL−1) was added to each well and the cells were incubated for another 4 h at 37 °C. The reaction products were then solubilized in 200 μL of DMSO before quantifying the color of reaction product using an ELISA reader (Multiscan MS, Labsystems, Helsinki, Finland) at 570 nm. Data were expressed as the percentage of viable control cells calculated from the absorbance at 570 nm by comparing the tests with the control.
Accelerated stability studies
Stability studies were carried out to determine the effect of accelerated storage conditions of temperature and humidity on the optimized NPs. The accelerated stability study was conducted according to International Conference on Harmonisation guidelines (ICH, Q1A (R 2) 2003; Shaikh et al. 2009). As per ICH guidelines Q1A (R 2), the accelerated stability study for the drug substances intended for storage in a refrigerator should be carried out at 25 ± 2 °C/60 ± 5 % RH. In this study, sealed vials of freshly prepared and freeze-dried NPs were placed in stability chamber maintained at 25 ± 2 °C and 60 ± 5 % RH. Samples were withdrawn at 0, 15, 30, 45, 60, and 90 days and then evaluated for particle size and DL.
Shelf life was determined as the time at which the 95 % one-sided confidence limit for the mean curve intersects the acceptance criterion of 90 % drug remaining. The data was evaluated using Sigmaplot ™ 10 software (Cranes Software International, Bangalore, India). Percentage drug remaining was plotted against time in months to determine the shelf life.
Results and discussion
All the 17 batches proposed by Box–Behnken experimental design yielded NPs and the results are displayed in Table 3. However, Table 4 gives the optimization constraints selected for each variable, while Table 5 gives the optimized formula generated by Design Expert® software showing the predicted and experimental values obtained.
Particle size and particle size distribution
The optimized NPs obtained were in the size range of 180–230 nm with a mean particle size of 205.61 ± 10.4 nm. PDI of the optimized NPs was less than 0.3 in all the batches. The maximum PDI value obtained was 0.243, while the minimum value was 0.078. PDI values less than 0.3 indicated uniform size distribution of NPs in all batches.
When determining the effect of independent factors on particle size (R 1), the model proposed the following polynomial equation:
where R 1 is the particle size of NPs, A is the amount of PCL used (mg), B is the conc. of Pluronic F-68 used (%), and C is the amount of triclosan used (mg).
From the polynomial equation for particle size, a positive sign represented a synergistic effect, while a negative sign indicated an antagonistic effect. Factor A (PCL) appeared to have more profound effect on particles size than factor B and C. As the PCL level increased, the particle size also increased dramatically. The concentration of the Pluronic F-68 used affected the particle size in opposite direction to that observed with factor A (PCL level). The negative coefficient value of factor B indicated the decrease in particle size with an increase in factor B (Pluronic F-68 concentration). It was observed that, when higher concentrations of Pluronic F-68 were used, smaller particle size was obtained. This might be due to the fact that higher amount of Pluronic stabilizes the formed NPs and prevented it from aggregation. The lowest particle size was obtained (169.5 nm) when 1 % Pluronic F-68 (maximum level of surfactant concentration) was used. Factor C (drug level) showed only slight positive effect on particle size as compared to that observed on factor A (PCL), which showed a larger positive effect. It was observed that, when higher level of triclosan was used, the particle size also increased but only slightly. The Model F value was 23.64, which implied that the model was significant. There was only a 0.02 % chance that a “Model F Value” this large could occur due to noise. The “Lack of Fit F value” of 2.24 implied that, the Lack of Fit was not significant (p value = 0.2262). Non-significant lack of fit is good, as the model is required to fit. Values of “Prob > F” (p value) less than 0.0500 indicated model terms were significant. In this case, A, B, and A 2 are significant model terms. A, B, and A 2 had all appeared to be significant models in such a way that the shift in the model values directly reflects in the response values. “Adeq Precision” was 17.370. “Adeq Precision” measures the signal-to-noise ratio. A ratio greater than 4 was desirable. This ratio of 17.370 indicated an adequate signal. Thus, this model could be used to navigate the design space. Figure 1a–d shows the effect of PCL levels on particles size. Figure 1a, b, e, and f shows the effect of Pluronic F-68 concentrations on particle size. Figure 1c–f shows the effects of drug levels on particle size.

Contour and response surface plots for particle size (R 1). a Contour plot of factor B versus A against R 1; b response surface plot of factor B versus A against R 1; c Contour plot of factor C versus A against R 1; d response surface plot of factor C versus A against R 1; e contour plot of factor C versus B against R 1; f response surface plot of factor C versus B against R 1
These results were in conformity as published by Piñón-Segundo et al. (2005) and associates who observed similar effects when triclosan-loaded polymeric NPs were prepared. It was observed that, the use of higher quantity of triclosan lead to the increase in NPs mean size.
Entrapment efficiency and DL
The percentage EE and percentage DL for the optimized nanoparticles were found to be 91.02 ± 2.4 and 21.71 ± 1.3 %, respectively.
On finding the effect of independent factors on percentage EE (R 2), the model proposed the following polynomial equation for EE:
where R 2 is the EE of drug loaded NPs, A is the amount of PCL used (mg), B is the conc. of Pluronic F-68 used (%), and C is the amount of triclosan used (mg).
As indicated from the polynomial equation above, increase in PCL level lead to the slight increase in EE. In this model, factor B appeared to have a negative effect on EE although the effects were not broad. Therefore, as the concentration of Pluronic F-68 increased, the EE value decreased slightly. This might be due to increased solubility of triclosan (Factor C) in higher concentration of Pluronic F-68, which might lead to washing out of triclosan that were entrapped on the NPs peripherial surfaces, and hence resulted in the decrease of EE. Factor C also showed some effects on the EE. It was observed that, the EE of triclosan in PCL NPs increased linearly with increasing drug concentration to a maximum of 94.2 %. The maximum EE obtained was as a result of the addition of 7.5 mg of triclosan. A plateau was observed at this point, and increasing the amount of triclosan did not resulted in any enhanced EE, but rather, declined EE slightly.
The model was found to be significant as the obtained F value was 31.79. The Lack of Fit was not significant (p value = 0.7085). The model terms B, C, and C 2 were found to be significant. The model “Pred R-Squared” was 0.8701. “Pred R-Squared” of 0.8701 was in reasonable agreement with the “Adj R-Squared” of 0.9454. “Adeq Precision” was 16.052 and thus indicated an adequate signal. Figure 2a–d shows the effect of PCL levels on EE. Figure 2a, b, e, and f shows the effect of Pluronic F-68 concentrations on EE. Figure 2c–f shows the effects of triclosan levels on EE.

Contour and response surface plots for EE (R 2). a Contour plot of factor B versus A against R 2; b response surface plot of factor B versus A against R 2; c contour plot of factor C versus A against R 2; d response surface plot of factor C versus A against R 2; e contour plot of factor C versus B against R 2; f response surface plot of factor C versus B against R 2
Similar results were obtained by Chawla and Amiji (2002) when they prepared tamoxifen-loaded PCL NPs. In another study, Zili and associates prepared griseofulvin-loaded PCL NPs and observed that, the percentages of NPs encapsulation efficiency are inversely proportional to griseofulvin concentration (Zili et al. 2005).
On determining the effect of independent factors on percentage DL (R 3).
The model proposed the following linear equation:
where R 3 is the DL of triclosan loaded NPs, A is the amount of PCL used (mg), B is the conc. of Pluronic F-68 used (%), and C is the amount of triclosan used (mg).
Factor A (PCL level) showed positive effect on DL as indicated by the equation above. It was observed that, when higher level of PCL was used, the DL also increases slightly. Factor B which was the concentration of the Pluronic F-68 used did not show any significant effect on DL. Only a small negligible effect was observed. The negative coefficient value of factor B indicated the decrease in DL with an increase in factor B. Factor C (drug level) appeared to have more profound effect on DL than other factors. As the triclosan level increased, the DL also increased linearly. The F value for this model was 42.45 and this showed that the model is significant. A p value of 0.1379 implied that, the Lack of Fit was not significant. Non-significant lack of fit is good, as the model is required to fit. Values of “Prob > F” (p value) less than 0.0500 indicated model terms were significant. In this case, A, C, and C2 are significant model terms. The model “Pred R-Squared” was 0.7864. “Pred R-Squared” of 0.7864 was in reasonable agreement with the “Adj R-Squared” of 0.9589. The adeq Precision of 23.139 indicated an adequate signal. Figure 3a–d shows the effect of PCL levels on DL. Figure 3a, b, e, and f shows the effect of Pluronic F-68 concentrations on DL. Figure 3c–f shows the effects of drug levels on DL. A similar effect was also observed be Piñón-Segundo et al. (2005). When they prepared triclosan-loaded polymeric NPs, it was observed that, the DL increased linearly with an increase in triclosan level.

Contour and response surface plots for DL (R 3). a Contour plot of factor B versus A against R 3; b response surface plot of factor B versus A against R 3; c contour plot of factor C versus A against R 3; d response surface plot of factor C versus A against R 3; e contour plot of factor C versus B against R 3; f response surface plot of factor C versus B against R 3
Zeta potential
Triclosan-loaded NPs exhibited a zeta potential of −33.8 ± 1.1 mV on zeta potential analyzer (Zetasizer Nano ZS, Malvern Instruments Ltd., Worcestershire, United Kingdom). It also showed zeta deviation of 5.58 ± 0.6 mV and conductivity of 0.0072 mS/cm. On the other hand, the zeta potential of triclosan-free NPs (placebo) was −7.32 ± 0.9 mV, with zeta deviation of 5.59 ± 0.5 mV and conductivity of 0.00857 mS/cm. The value of zeta potential of the NPs increased in magnitude when triclosan was loaded. The surface charge density of the NPs was probably increased due to the preferential surface localization of triclosan on NPs. Surface localization of triclosan was later confirmed by the burst release observed in the in vitro release studies. Similar results were observed by Chawla and Amiji (2002) when they prepared and characterized tamoxifen-loaded PCL NPs.
SEM and TEM
The SEM results indicated that all NPs had spherical solid matrix structures and smooth surface. The images of the optimized NPs are shown in Fig. 4. Figure 4a showed one of the largest nanoparticle formed which could be as a result of higher drug load. Figure 4b showed the dried triclosan-loaded NPs with homogenous spherical shape and without any evidence of crystals on the surface of NPs, while Fig. 4c showed NPs with the highest concentration of triclosan, which indicated that high triclosan ratio leads to formation of fusion and film like structure. This indicated that triclosan also possess plasticity effects on PCL.

SEM images of the optimized NPs: a Single nanoparticle displayed at magnification of greater than ×14,000 b NPs displayed at magnification of ×13,400 c NPs displayed at magnification of less than ×10,000
The particles size ranges of the optimized NPs obtained with particle size analyzer, Nano ZS Zetasizer were confirmed by TEM analysis. The test gives the size ranges 180–230 nm as shown in Fig. 5. The NPs were found to be spherical and homogenous.

TEM image of optimized NPs
DSC
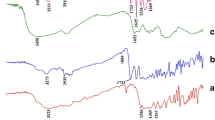
Figure 6 showed the DSC thermograms of free triclosan, pure PCL, and triclosan-loaded NPs, respectively. Free triclosan showed an endothermic peak of melting at 64.02 °C as indicated in Fig. 6a, while the pure PCL showed it endothermic peak of melting at 65.94 °C (Fig. 6b). Triclosan-loaded PCL NPs showed its endothermic peak of melting at 54.09 °C (Fig. 6c). DSC studies did not detect any crystalline drug material in the nanoparticle samples which might be due to change of crystalline form to amorphous form, and this confirmed the molecular level dispersion of triclosan. After the DSC process, triclosan could be in an amorphous or disordered-crystalline phase of a molecular dispersion or a solid solution state in the polymer matrix (Mu and Feng 2002). Kim et al. (2006) and associates obtained similar results when they prepared and characterized chitosan/gelatin microcapsules containing triclosan.

DSC thermograms: a DSC thermogram of free triclosan; b DSC thermogram of pure PCL; c DSC thermogram of triclosan-loaded PCL NPs
In vitro drug release studies
In the release profile of triclosan, shown in Fig. 7, there was an initial burst release effect as about 76 % of the entrapped drug was released in the first hour. The maximum amount of triclosan was released within a time of about 3 h. The burst release effects of hydrophobic drugs like triclosan may be attributed to the predominant surface entrapment of the drug on NPs. Similar release profile was observed for triclosan in the study conducted by Pinon-Segundo and coworkers. In their study, in which triclosan-loaded PLGA NPs were also in direct contact with the dissolution medium (2 % w/v Brij® 58 aqueous solution), they observed that, about 75 % of the entrapped drug was released within 30 min (Piñón-Segundo et al. 2005). The rapid burst effect of triclosan might be attributed to higher localization of the drug on the surface of the NPs. The burst release observed may also be attributed to the low molecular weight of the drug and the smaller size of the particles (Huang and Brazel 2001; Chawla and Amiji 2002).

In vitro release profile of triclosan from PCL NPs. (n = 3)
Cell viability assay
The results of the cell viability studies are presented in Table 6 and Fig. 8. The cell viabilities were expressed as percentage of the control group. From the data obtained from the cell viabilities of L-929 cell lines treated with different samples, it was clear that the mean cell viability shown with triclosan NPs were very close to that of control (i.e., 115.66 % of the control). This means that the viability of cells was not affected by the presence of triclosan loaded nanoparticles. Thus, the percentage cell viability values (with respect to control group) near to 100 indicated that the triclosan loaded nanoparticles were not cytotoxic to the cells. Thus, it could be inferred that the triclosan-loaded polycaprolactone nanoparticles would not cause any toxicity to the periodontal tissues during its use. The cell viability values of triclosan alone and placebo nanoparticles were also very much comparable with that for control group. The mean cell viability values observed were 108.52 and 109.62 for triclosan alone and placebo nanoparticles, respectively. This result indicated that neither triclosan nor polycaprolactone used for the fabrication of nanoparticles would cause any toxicity on periodontal tissue during use. The results of cell viability expressed as bar diagrams (Fig. 8) give a better understanding of the observed results. From the figure it is very clear that the cell viability values of all groups were comparable to the control group.

Cell viabilities of L-929 cell lines treated with different samples
Accelerated stability studies
Up to 98.46 % of the drug content in NPs remained after the period of 90 days of storage at 25 ± 2 °C and 60 ± 5 % RH. The observed parameters, i.e., particle size and DL were analyzed for statistical significance by one way analysis of variance (ANOVA) followed by Tukey–Kramer multiple comparison test using Prism® 5 software (GraphPad Software, Inc., CA, USA). The changes in the observed factors were not statistically significant (p > 0.05) which indicated that the optimized nanoparticulate formulation was stable. The results are given in Table 7. The shelf life was found to be 17.07 months using the Sigmaplot ™ 10 software (Fig. 9).

Percentage triclosan remaining versus time (months) plot for shelf life determination of optimized triclosan-loaded PCL NPs
Conclusions
We synthesized and optimized triclosan-loaded PCL nanoparticles. Optimization of the triclosan-loaded PCL nanoparticle formulations was performed using Box–Behnken design. The derived polynomial equations and model plots (i.e., contour and response surface plots) aided in predicting the values of selected independent variables for the preparation of optimized nanoparticle formulations with desired properties. The optimized nanoparticles were evaluated for its particle size, particle size distribution, zeta potential, surface morphology, in vitro drug release, and thermal properties. Cell viability assay was also carried out with the optimized nanoparticles. The results indicated the absence of cytotoxicity of the triclosan loaded NPs. The nanoparticles were found to be stable as revealed from the accelerated stability studies. The optimized nanoparticles were having sufficient shelf life. On the basis of these research findings, it was concluded that triclosan-loaded PCL NPs were developed and optimized which could serve as a novel colloidal drug delivery system for the treatment of periodontal infections.
References
Bollen CM, Quirynen M (1996) Microbiological response to mechanical treatment in combination with adjunctive therapy. A review of the literature. J Periodontol 67:1143–1158
Chawla JS, Amiji MM (2002) Biodegradable poly(ε-caprolactone) nanoparticles for tumor-targeted delivery of tamoxifen. Int J Pharm 249:127–138
Chiappetta DA, Degrossi J, Teves S, D’Aquino M, Bregni C, Sosnik A (2008) Triclosan-loaded poloxamine micelles for enhanced topical antibacterial activity against biofilm. Eur J Pharm Biopharm 69:535–545
Espuelas MS, Legrand P, Irache JM, Gamazo C, Orecchioni AM, Devissaguet JP, Ygartua P (1997) Poly(ε-caprolactone) nanospheres as an alternative way to reduce amphotericin-B toxicity. Int J Pharm 158:19–27
Huang X, Brazel CS (2001) On the importance and mechanisms of burst release in matrix-controlled drug delivery systems. J Control Release 73:121–136
International Conference on Harmonisation (2003) ICH Harmonised Tripartite Guideline: Stability testing of new drug substances and products, Q1A(R2). International Conference on Harmonisation, Geneva
Jain N, Jain GK, Javed S, Iqbal Z, Talegaonkar S, Ahmad FJ, Khar RK (2008) Recent approaches for the treatment of periodontitis. Drug Discov Today 13:932–943
Kim JC, Lee HY, Kim MH, Lee HJ, Kang HY, Kim SM (2006) Preparation and characterization of chitosan/gelatin microcapsules containing triclosan. Colloids Surf B 52:52–56
Koleske JV (1978) Blends containing poly(ε-caprolactone) and related polymers. In: Paul DR, Newman S (eds) Polymer blends, vol 2. Academic Press, New York, pp 369–389
Medlicott NJ, Rathbone MJ, Tucker IG, Holborow DW (1994) Delivery systems for the administration of drugs to the periodontal pocket. Adv Drug Deliv Rev 13:181–203
Mu L, Feng SS (2002) Vitamin E TPGS used as emulsifier in the solvent evaporation/extraction technique for fabrication of polymeric nanospheres for controlled release of paclitaxel (Taxol®). J Control Release 80:129–144
Murthy RSR (1997) Biodegradable polymers. In: Jain NK (ed) Controlled and novel drug delivery. CBS Publisher, New Delhi, pp 27–51
Ozmeric N (2004) Advances in periodontal disease markers. Clin Chim Acta 343:1–16
Pihlstrom BL, Michalowicz BS, Johnson NW (2005) Periodontal diseases. Lancet 366:1809–1820
Piñón-Segundo E, Ganem-Quintanar A, Alonso-Pérez V, Quintanar-Guerrero D (2005) Preparation and characterization of triclosan nanoparticles for periodontal treatment. Int J Pharm 294:217–232
Quintanar-Guerrero D, Allemann E, Fessi H, Doelker E (1998) Preparation techniques and mechanisms of formation of biodegradable nanoparticles from preformed polymers. Drug Dev Ind Pharm 24:1113–1127
Rosling B, Dahlen G, Volpe AR, Furuichi Y, Ramberg P, Lindhe J (1997) Effect of triclosan on the subgingival microbiota of periodontitis-susceptible subjects. J Clin Periodontol 24:881–887
Schroeder HE (1976) Pathogenesis of inflammatory periodontal disease: a summary of current work. Lab Invest 34:235–249
Schwach-Abdellaoui K, Vivien-Castioni N, Gurny R (2000) Local delivery of antimicrobial agents for the treatment of periodontal diseases. Eur J Pharm Biopharm 50:83–99
Shaikh J, Ankola DD, Beniwal V, Singh D, Ravi-Kumar MNV (2009) Nanoparticle encapsulation improves oral bioavailability of curcumin by at least ninefold when compared to curcumin administered with piperine as absorption enhancer. Eur J Pharm Sci 37:223–230
Sinha VR, Bansal K, Kaushik R, Kumria R, Trehan A (2004) Poly-ε-caprolactone microspheres and nanospheres: an overview. Int J Pharm 278:1–23
Slots J, Rams TE (1990) Antibiotics in periodontal therapy: advantages and disadvantages. J Clin Periodontol 17:479–493
Southard GL, Godowski KC (1998) Subgingival controlled release of antimicrobial agents in the treatment of periodontal disease. Int J Antimicrob Agents 9:239–253
Zili Z, Sfar S, Fessi H (2005) Preparation and characterization of poly-ε-caprolactone nanoparticles containing griseofulvin. Int J Pharm 29:261–267
Acknowledgments
Authors would like to thank the staff of Electron Microscopy Dept., AIIMS for conducting SEM and TEM analysis. Mr. Nafiu Aminu (M. Pharm.) thanks Zamfara state government of Nigeria for its inestimable and incessant support to him in this project.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Aminu, N., Baboota, S., Pramod, K. et al. Development and evaluation of triclosan loaded poly-ε-caprolactone nanoparticulate system for the treatment of periodontal infections. J Nanopart Res 15, 2075 (2013). https://doi.org/10.1007/s11051-013-2075-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-013-2075-6