
Abstract
The electronics industry is one of the world’s fastest growing manufacturing industries. However, e-waste has become a serious pollution problem. This study reports the recovery of e-waste for preparing valuable MCM-48 and ordered mesoporous carbon for the first time. Specifically, this study adopts an alkali-extracted method to obtain sodium silicate precursors from electronic packaging resin ash. The influence of synthesis variables such as gelation pH, neutral/cationic surfactant ratio, hydrothermal treatment temperature, and calcination temperature on the mesophase of MCM-48 materials is investigated. Experimental results confirm that well-ordered cubic MCM-48 materials were synthesized in strongly acidic and strongly basic media. The resulting mesoporous silica had a high surface area of 1,317 m2/g, mean pore size of about 3.0 nm, and a high purity of 99.87 wt%. Ordered mesoporous carbon with high surface area (1,715 m2/g) and uniform pore size of CMK-1 type was successfully prepared by impregnating MCM-48 template using the resin waste. The carbon structure was sensitive to the sulfuric acid concentration and carbonization temperature. Converting e-waste into MCM-48 materials not only eliminates the disposal problem of e-waste, but also transforms industrial waste into a useful nanomaterial.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Silica-based mesoporous materials, designated M41s, have attracted much attention because these materials exhibit a high surface area, large pore volume, and adjustable pore size. Mesoporous silica with pore sizes ranging from 2 to 50 nm has found many applications, including catalysts (Ying et al. 2011), nanocomposites (Kim et al. 2009), storage and recovery of H2 and CO2 (Prasanth et al. 2010; Jang et al. 2009), and biotechnological applications (Liu et al. 2010). The main members of the M41’s family are MCM-41, MCM-48, and MCM-50, which have hexagonal, cubic, and unstable lamellar structures, respectively. Because MCM-48 material has a three-dimensional pore system that helps reduce pore blockage and provides more efficient mass transfer kinetics, it is a better candidate for catalytic and adsorption applications than with MCM-41 materials (Zhao et al. 2010a). The channel structure of MCM-48 silica consists of an interconnected pore system that acts as a template for ordered mesoporous carbon (OMC) synthesis (Ryoo et al. 2001). The ordered mesoporous carbon with large adsorption capacity is highly favorable for the development of advanced materials in CH4 and H2 storage (Peng et al. 2008), adsorbents for removal of organic pollutants (Hao et al. 2010), carbon composite films for supercapacitor (Dai et al. 2011), fuel cells (Salgado et al. 2010), and biomedical materials (Kim et al. 2008). However, owing to its very sensitive synthetic conditions, relatively few studies investigate MCM-48. Electronic packaging resin waste is rich in silica. Thus, achieving high-quality MCM-48 and OMC materials with well-ordered mesoporous structure by a green approach and electronic waste has important economic and environmental implications.
Over the past decades, the electronics industry has developed rapidly around the world. Estimations of the worldwide revenue of the semiconductor industry exceeded US 226 billion in 2009, and this figure is expected to grow by 13 % each year (Kim et al. 2010). However, the disposal of electronic and electrical waste (e-waste) has become a serious environmental problem as a result of the huge amounts of electronic goods being produced. The current annual production of e-waste, including old computers, mobile phones, television sets, stereos, and radios, is estimated to be approximately 20–50 million tons (Chen et al. 2011). Taiwan’s Environmental Protection Administration (EPA) established its Scrap Home Appliances and Computers Recycling Regulation, based on the Waste Disposal Act (WDA), to require manufacturers and importers to take back their products (Shih 2001). Electronic packaging materials are widely used in many types of electrical and electronic equipment. This material is used to protect electronic products such as integrated circuits (IC). Electronic packaging material is generally a mixture of epoxy resin, phenol resin, silica, and additives (Liou 2003). In present, the treatment of e-waste includes reuse, remanufacturing, and recycling, along with incineration and landfilling (Cui and Zhang 2008). Because plastic materials are essentially non-biodegradable, putting them in landfill is not a viable long-term solution. The chemical composition of packaging resin waste is approximately 80 wt% silica (Liou 2004), which is useful for the mass production of MCM-48 mesoporous molecular sieves. Therefore, using recycled industrial resin waste offers benefits in terms of waste treatment and the recovery of valuable materials.
Numerous studies indicate the synthesis and characterization of mesoporous MCM-41 materials by adding an appropriate surfactant to the silica precursor (Bhagiyalakshmi et al. 2010a; Lelong et al. 2008). However, the use of e-waste to prepare MCM-48 materials is generally neglected in the literature. The phase characteristics of MCM-48 are strongly influenced by its synthesis time, temperature, silica source, and type of surfactant template (Peña et al. 2001; Petitto et al. 2005). Doyle et al. (2006) studied the evolution of phases during MCM-48 synthesis and found that increasing reaction time or temperature alters the overall phase transformations from MCM-41/MCM-50 to MCM-48 to MCM-50. Wei et al. (2010) synthesized MCM-48 with mixed surfactants of cethyltrimethylammonium bromide (CTAB) and Triton X-100 (TX-100), showing that TX-100/CTAB ratio is crucial to the formation of a pure MCM-48 structure. Solid wastes, such as coal fly ash and rice husk, provided the silicon sources for the synthesis of mesoporous silica materials, SBA-15 and SBA-16 (Chandrasekar and Ahn 2008; Bhagiyalakshmi et al. 2010b). These mesoporous silica materials were impregnated with sucrose in the presence of sulfuric acid solution and then were pyrolyzed in inert gas to obtain ordered mesoporous carbon.
The recovery of valuable metals from e-waste has been well studied in the literature (Cui and Zhang 2008). However, little attention has been paid to the recovery and utilization of silica from e-waste. The current study investigates MCM-48 prepared using a sodium silicate precursor obtained from recycled electronic packaging resin wastes. The commercial fabrication of sodium silicate from quartz sand and sodium carbonate at 1,300 °C requires a lot of energy (Brinker and Scherer 1990). In contrast, using cheap inorganic silica sources from e-wastes can improve the industrial scale production of MCM-48 materials. Previous studies provide a detailed analysis of the recovery of packaging resin ash and its application in preparing MCM-41 materials (Liou 2003; 2004; 2011). The aim of this study is to synthesize highly ordered MCM-48 using cationic and neutral surfactant mixture. The high-quality MCM-48 produced in this study is suitable as a template for the synthesis of ordered mesoporous carbons. The synthesis of MCM-48 and ordered mesoporous carbon by a green approach enables the effective use of e-waste resources.
Experimental
Materials
The electronic packaging resin waste used in this study was obtained from an electronic packaging mill (Siliconwave Precision Co., Taiwan). The basic constituents and properties of this material were described in a previous report (Liou 2003). Commercially available surfactants, including cetyltrimethylammonium bromide (CTAB, C16H33(CH3)3NBr, Acros organics) and polyethylene glycol dodecyl ether (C12(EO)4, Sigma-Aldrich), served as organic templates. Analytical grade sulfuric acid (H2SO4), hydrochloric acid (HCl), hydrofluoric acid (HF), and sodium hydroxide (NaOH) were used as acid and base treatments (Merck, Germany). Sucrose (C12H22O11) and glucose (C6H12O6) were used as the carbon precursors (Sigma-Aldrich). High purity air and N2 were used as the reaction gas and purge gas, respectively (Sun Fu Co., 99.995 %).
Extraction of silica from electronic packaging resin waste
Resin wastes were first ground and fine powders were obtained by sieving to below 45 μm. The packaging resin ash (denoted as PRA) was used as a raw material for preparing mesoporous silica by Liou’s method (Liou 2004). For each heat-treated sample, the powdered resin of approximately 5.0 g was heated at 800 °C in air. The amount of ash produced is 4.1 g. After the heating period, the ash was refluxed using HCl solution to remove any metallic impurities. The remaining solid was washed with distilled water several times to remove acid from the filtrate, and then dried in an oven at 105 °C for 24 h.
The dissolution of silica from PRA was performed by an alkali-extracting method (Liou 2011). The 50 g of acid-leached PRA powder was added to a flask containing a 4.0 M 325 ml of NaOH solution and boiled at 100 °C for 6 h with constant stirring. This process converted the silica into a sodium silicate solution. The solution was centrifuged to separate the suspended particles, and then filtered through a glass filter (Whatman plc, England) to remove any metal or carbon residue. This filtration process was repeated several times and finally obtained a colorless, transparent solution. The concentration of alkali-extracting solution was diluted with deionized water from 4.0 M to 2.0 M.
Synthesis of mesoporous silica molecular sieves
By a typical synthesis procedure, 1.82 g of CTAB and 0.27 g of C12(EO)4 were dissolved in 32 ml of deionized water. The 10 ml of sodium silicate solution was mixed with the CTAB/C12(EO)4 solution with constantly stirring. A 1.0 M H2SO4 solution was titrated slowly into the silicate solution. The mixture pH was monitored with a S20-K pH meter (Metter Toledo) to control pH at a constant value of 1–11. The gel was transferred into PTFE-lined stainless steel autoclave and allowed to hatch at a constant temperature for 48 h. The precipitates were water-washed, filtered, and dried. The dried powders were calcined in air at a 1 °C/min heating rate and then maintained at 450–850 °C for 6 h.
Preparation of mesoporous carbon molecular sieve
The OMC synthesis method reported by Ryoo et al. (2001) was adopted. Essentially, MCM-48 was impregnated with sucrose and water in the presence of H2SO4. The impregnated silica was dried in an oven at 100 °C for 6 h, and then dried at 160 °C for 6 h. The impregnation and drying steps were repeated once. The obtained sample was placed in a tubular reactor and was heated to 500–900 °C at a temperature rate of 10 °C/min in N2. After carbonization, the silica template was removed in an aqueous HF solution. Finally, the OMC was obtained by washing the solid sample repeatedly with deionized water and drying it in air at 105 °C for 24 h.
Characterization of the mesoporous silica and mesoporous carbon samples
The amounts of metallic impurities were estimated using an inductively coupled plasma-mass spectrometer (Kontrin Plasmakon, model S-35). A Heraeus elemental analyzer was used to determine the amount of fundamental organic element. The adsorption–desorption isotherms of N2 at −196 °C were measured using a Micrometric ASAP 2010 instrument.
The mesophase of samples was analyzed using a small angle X-ray scattering (SAXS) system (Osmic, model PSAXS-USH-WAXS-002). The crystalline structure of the samples was identified using powder X-ray diffractometer (PANalytical, model X’pert pro system). The FTIR spectra of samples were obtained using a Shimadzu FTIR-8300 spectrometer. The surface morphology of samples was studied using a JEOL JSM-6700F field-emission scanning-electronic microscope (FE-SEM). The mesoporous structure of powders was observed by high-resolution transmission electron microscopy (JEOL, model JEM-1200CX II). The remaining amount (W/W o) of samples were recorded using a thermogravimetric apparatus (Mettler, model TGA/SDTA851e). Each sample was heated at a 10 °C/min heating rate in air.
Results and discussion
Analysis of metallic impurities of silica molecular sieve
The amounts of metallic ingredients present in PRA and mesoporous silica samples were analyzed by ICP-MS (Table 1). When silica was synthesized from alkali-extracted PRA at pH 7 without the addition of a surfactant, the sample possessed a higher proportion of Na impurities. This is because Na salts are embedded in the silica gel during precipitation and are not easily removed from the gel (Liou 2011). No Na impurities appeared in the mesoporous silica samples, which were synthesized from alkali-extracted PRA by adding a surfactant. This is due to the well-dispersed silica particles in the surfactant solution. The Na ions can easily diffuse out of the silica matrix and be removed by a water-washing procedure. Comparing silica samples synthesized at various pH values shows that the total impurity content produced under strongly acidic (pH 1) and strongly basic (pH 11) conditions is relatively lower than neutral (pH 7) and weakly acidic (pH 3 and pH 5) conditions. The silica synthesized at pH 5 has highest impurity content. The remaining masses for thermal decomposition of as-synthesized silica in air are in the order of pH 5>pH 3>pH 7>pH 1>pH 9>pH 11 (in subsequent TGA observations). This suggests that when silica materials are synthesized in the neutral and weakly acidic media, it results in a higher degree of poly-condensation of the silicate species. As a result, trace amounts of metal may be cached in the silica matrix and cannot be removed by a water-washing procedure. The purity of PRA-MCM-48 silica synthesized at pH 11 is as high as 99.87 wt%.
Synthesis conditions of silica molecular sieve
Effects of gelation pH value
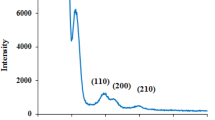
Figure 1a shows that a maximum surface area of 1,153 m2/g at pH 11. According to SAXS analysis (Fig. 1c, d), the three diffraction peaks at (211), (220), and (332) appeared when the solution pH was adjusted to pH 1 or pH 11. The two SAXS patterns correspond to the characteristics of the MCM-48 material (Han et al. 2004; Doyle and Hodnett 2003). This result can be explained by the electrostatic attraction between charged silica species and surfactant micelles (Zhao et al. 2010b). At a high pH (pH 11), the negatively charged silica interacted with positively charged CTA+ micelles, leading to the formation of a cubic framework. Therefore, the silica possessed a high surface area. At a low pH (pH 1), an ordered mesophase was constructed through reversal arrangement of cationic silica and neutral micelles (Liou 2011). This situation also increases the surface area of the products. At pH 3–7, the SAXS patterns show mainly one diffraction peak at 1.26°. This indicates that the surfactant micelle may be destroyed, converting the pore structure into a less ordered structure. Consequently, the surface area steeply decreases. The SAXS pattern for pH 9 sample shows an intense diffraction (100) peak and three higher order (110), (200), and (210) reflections at 2Θ values below 10°, indicating the hexagonal arrangement of a MCM-41 mesoporous structure (Samadi-Maybodi et al. 2011). Figure 1b shows that the silica yield has the opposite tendency as surface area analysis. The silica yield from PRA was defined as (weight of mesoporous silica produced after calcination)/(weight of PRA sample) × 100. For further comparison, Table 2 shows the relationship between gelation pH and the amount of organic element in the as-synthesized samples. The silica phase at pH 5 is approximately 82 wt%, and at pH 1 and pH 11 has wt% only around 64 and 40, respectively. These observations indicate that the higher proportion of silicate species in the surfactant solution under neutral and weakly acidic conditions increases the silica yield for pH 7 and pH 5 samples.

Effect of gelation pH values on a surface area, b silica yield, and c, d phase characteristic of mesoporous silica samples
Effects of neutral/cationic surfactant ratio
Figure 2a shows the surface area of mesoporous silica obtained by changing the neutral-to-cationic surfactant ratio, where a fraction of the neutral surfactant, f n, varied from 0 to 1. The highest surface area of 1,317 m2/g appeared at a fraction of 0.04. Based on SAXS observations (does not show here), typical hexagonal phase MCM-41 products appeared without the addition of neutral surfactant (f n = 0) (Ryoo et al. 1999). A pure mesophase MCM-48 was obtained as f n increased to 0.04 and 0.13. When the f n was 0.26, the product was lamellar. Further increasing f n beyond 0.40, the mesophase transformed to a poor structural order. Therefore, the surface area steeply reduced. This observation indicates that excessive neutral surfactant can dilute the CTA+ concentration, causing the micelles to gradually disappear and transforming the lamellar phase to the disordered phase.

Effect of a neutral/cationic surfactant ratio, b hydrothermal treatment temperature, and c calcination temperature on the surface area of mesoporous silica samples synthesized at pH 11
Effects of hydrothermal treatment temperature
Figure 2b shows that the surface area increases with increasing hydrothermal treatment temperature from 25 to 75 °C is due to the progressive formation of ordered mesophase silica. However, the surface area decreases with increasing temperature from 75 to 150 °C due to silica pore shrinkage. This suggests that increasing the hydrothermal treatment temperature can accelerate the silicate condensation on the silica wall, which subsequently thickens the silica framework (Wang et al. 2000). Mesoporous silica obtained at a low temperature (25–75 °C) has the characteristics of a hexagonal MCM-41 structure. At high temperature (100–125 °C), the silica shows the characteristics of a cubic MCM-48 structure. When the hydrothermal temperature is 150 °C, the material structure transforms to the disordered phase accompanied by a significant decrease in surface area.
Effects of calcination temperature
The purpose of calcination is to remove the organic template from silica/surfactant composite, leaving the porous surface of product. Figure 2c shows that the maximum of the BET surface area appears at 550 °C. The surface areas increase as the calcination temperature increases from 450 to 550 °C due to the release of organic volatiles. However, the surface areas decrease when the calcination temperature exceeds 550 °C is probably caused by the collapse of the gel structure due to the rapid release of water from the pores.
Analysis of silica product characteristics
Pore structure analysis of mesoporous silica molecular sieve
Figure 3 shows the N2 adsorption–desorption isotherms of silica samples. The isotherms for samples synthesized at pH 1–5 (Fig. 3a) show a type IV isotherm, typical characteristics of mesoporous solids. The sample at pH 5 shows a hysteresis loop similar to H3 (Sing et al. 1985). The isotherm for the pH 3 sample is similar to that of the pH 5 sample. The sample synthesized at pH 1 shows a type H2 hysteresis loop and an obvious increase in adsorbed volume. Figure 3b shows the pore size distribution of silica samples produced in acidic conditions. The silica produced at pH 1 shows a narrow pore distribution with average size of 3.7 nm. The samples synthesized at pH 3 and pH 5 have a wide pore size distribution and larger pore diameter, showing that the pore structure is not uniform.

Nitrogen adsorption–desorption isotherm and differential pore size distribution of mesoporous silica samples synthesized at various pH values: a, b pH 1–5; c, d pH 7–11
The isotherms of samples obtained at pH 7–11 are type IV (Fig. 3c). These pores may come from the primary pores of silica framework and the voids between particles (Han et al. 2004). The isotherm of the sample synthesized at pH 7 shows a hysteresis loop similar to H3. The pH 9 and pH 11 samples show a sharp increase in adsorbed volume at P/P o = 0.2–0.4 due to the capillary condensation of nitrogen, implying a uniform size of the pore channels (Liu et al. 2009). One H2 hysteresis loop appears at 0.4 < P/P o < 1.0 due to the formation of a mesoporous structure. The pore size distribution of silica samples synthesized at pH 7–11 is shown in Fig. 3d. The maximum pore distribution ranges from 2.5 to 3.1 nm increases with pH.
Table 3 lists the specific surface area, pore volume, and average pore diameter of PRA and mesoporous silica samples. In acidic media (pH 1–5), the surface area and pore volume decreased with the increase of the pH values. In neutral and basic media (pH 7–11), the surface area increased and pore volume decreased with the increase of the pH value. The maximum pore volume was 0.985 cm3/g at pH 7 and the minimum pore volume was 0.444 cm3/g at pH 5.
Analysis of thermal characteristic, chemical constitution, and crystalline structure
Figure 4a shows the TG and DTG curves of as-synthesized samples obtained in acidic media. The mass loss occurs in two temperature zones, revealing an initial mass loss for temperatures up to ~400 °C due to the decomposition and thermodesorption of the cationic surfactant. The first peak temperature at approximately 228 °C is related to the Hoffmann elimination (giving alkene and trimethylamine) and subsequent trimethylamine desorption (Zhao et al. 2005). The second and third peaks at approximately 275 and 330 °C can be associated with further thermal decomposition of surfactant intermediates. The mass loss increased as the pH decreased. This is because the content of organic substance in the as-synthesized samples increased with decreasing the pH values (Table 2). The mass loss in the range of 400–800 °C may be attributed to the release of water via condensation of silanol group (Kruk et al. 2000). Figure 4b shows the TG and DTG curves of as-synthesized samples produced in neutral and basic media. This figure reveals two major mass loss regions: ~330 °C, 330–800 °C. The DTG curves reveal two peak temperatures at 179–249 and 305 °C for pH 7–9. This mass loss is due to the decomposition and desorption of the organic templates and the water loss due to the condensation of silanols in the silica framework. The mass loss of sample increased as the pH increased because the content of organic substance in the as-synthesized sample increased.

TG and DTG thermograms of as-synthesized silica samples at various pH values: a pH 1–5; and b pH 7–11
Figure 5a shows the FTIR spectra of the MCM-48 silica obtained from PRA at various calcination temperatures. The bands appearing between 450 and 1,300 cm−1 are due to the presence of silica (Liou and Wu 2010). The bands at approximately 475 cm−1 correspond to the Si–O–Si vibrations. The peak at 970 cm−1 is related to the Si–OH bond, while the peak at 1,115 cm−1 is related to Si–O stretching vibrations. Surface silanols and adsorbed water molecules create the broad band appearing in the region between 3,200–3,700 cm−1. The peak position does not change when the temperature exceeds 550 °C, indicating that the combustion reaction is complete. Figure 5b shows the XRD pattern of MCM-48 with a wide range scan, revealing only a broad peak in the region 2Θ greater than 10o, which suggests the presence of disordered cristabolite (Liou and Yang 2011).

MCM-48 samples synthesized at pH 11: a Fourier transform infrared spectra obtained at various calcination temperatures; and b X-ray diffraction pattern obtained at calcination temperature of 550 °C
Synthesis of ordered mesoporous carbon molecular sieve
Experimental results indicate that the optimal procedure for synthesizing MCM-48 is to employ a neutral-to-cationic surfactant ratio of 0.04–0.13 at pH 11 with a hydrothermal temperature of 100 °C and a calcination temperature of 550 °C. Therefore, this study conducts the remaining experiments for preparing a MCM-48 template by these reaction conditions.
Figure 6 displays the SAXS patterns for carbons prepared by impregnation of the MCM-48 template with sucrose. As shown in Fig. 6a, the carbon sample obtained without the addition of sulfuric acid (H2SO4 = 0 g) did not have an ordered porous structure. However, adding varying amounts of sulfuric acid obtained CMK-1 carbons predominantly containing two sharp low-angle reflections typical of cubic structure (Hussain and Ihm 2009). This was caused by sucrose’s hydrophilic nature; it does not easily enter pores surrounded by a hydrophobic silica framework (Joo et al. 2001). However, adding a sulfuric acid catalyst transformed the sucrose into a less hydrophilic substance. As a result, the sucrose successfully penetrated the silica pore. When the amount of sulfuric acid was higher than 0.28 g (for each gram of sucrose), sulfuric acid occupying proportions of the silica pore volume may have caused the decreased SAXS intensity. Some carbons also formed outside the silica pores. Figure 6b shows the carbon sample obtained at 500 °C demonstrated weak diffraction intensities, indicating a poorly ordered mesoporous structure. When carbonization temperatures were in the range of 600 to 1,000 °C, these samples revealed characteristics of ordered mesoporous carbon with CMK-1 structure. The relative intensities of the diffraction peaks increased with increasing carbonization temperatures. Higher carbonization temperature facilitates the formation of carbons with enhanced framework and structural ordering. However, at a carbonization temperature of 1,000 °C, contraction or collapse of pores may have caused the resulting decrease in SAXS intensity (Liou and Wu 2009).

SAXS pattern for carbons synthesized by: a various amounts of H2SO4 for each grain of sucrose; and b various carbonization temperatures
Following carbonization of the sucrose/MCM-48 composites, removal of the MCM-48 templates by leaching using HF solution obtained pure mesoporous carbon products. The TG curves in Fig. 7 shows the effects of HF concentration on the amounts of silica template remaining after burning samples in air. The remaining silica decreased as the HF concentration increased. HF concentration of 5 wt% was sufficient for complete removal of the MCM-48 template. As revealed in the DTG curves in Fig. 7, the peak height increased with increasing HF concentrations, indicating that leaching of silica templates with high HF concentrations had the effect of increasing oxidation.

Effect of HF concentration on the remaining amount of ash for carbon sample burnt in air. The carbon samples were obtained from pyrolysis of sucrose/MCM-48 composite in N2 at 900 °C
Figure 8a, b show the N2 adsorption–desorption isotherm and pore size distribution for carbon obtained without the addition of sulfuric acid. The isotherm displayed the carbon sample including both microporous and mesoporous structures (Liou 2010). A narrow peak in pore size distribution occurred at 3.9 nm. The carbon sample exhibited a low BET surface area of approximately 877 m2/g and a total pore volume of 0.680 cm3/g. Figure 8c shows the N2 adsorption–desorption isotherm of CMK-1 carbon with carbonization temperature of 900 °C. The isotherm exhibits two capillary condensation steps at P/P o = 0.2–0.3 and 0.4–1.0. These well-pronounced capillary condensation steps indicate a high uniformity in pore size. The pore size distribution displayed in Fig. 8d reveals a bimodal mesoporous structure. Two narrow peaks appear at a pore diameter of approximately 2.3 and 3.9 nm. The CMK-1 sample exhibits a high BET surface area of 1,715 m2/g and total pore volume of 1.02 cm3/g.

Nitrogen adsorption–desorption isotherm and differential pore size distribution of mesoporous carbon samples prepared by the impregnation of MCM-48 template with sucrose: a, b without the addition of sulfuric acid; c, d addition of sulfuric acid
Observation of surface morphology of silica and carbon products
Figure 9a–g shows the surface morphologies of mesoporous silica obtained at pH 1–11. The silica sample synthesized at pH 1 consists of very fine grains with a mean size smaller than 5 nm, while the silica synthesized at pH 3 has a like-sponge porous tissue. The silica synthesized at pH 5 shows semi-sphere grains, while that formed at pH 7 shows an aggregate of fine grains. The MCM-41 sample synthesized at pH 9 shows irregularly shaped particles. The surface morphology of MCM-48 synthesized at pH 11 shows mainly rectangular particle shapes (Sayari 2000). Figure 9g shows the lamellar structure of silica synthesized at a neutral/cationic ratio of 0.26. The micrometer-sized spherical particles consist chiefly of thin platelets measuring 20-nm thick and 0.5-μm wide. Figure 9h–l shows the surface morphologies of mesoporous carbon samples. Figure 9h illustrates the direct synthesis of mesoporous carbon from as-synthesized MCM-48 silica nanocomposite. The proposed method uses CTAB/C12(EO)4 surfactants as a carbon precursor to substitute sucrose. SEM analysis shows that this carbon material is a hollow and spherical solid. SAXS confirms that this carbon material did not have an ordered porous structure (data not shown here). This observation indicates that the surfactant mixtures were unable to completely penetrate the silica pores. The surface area of direct synthesis of carbon material is only 750 m2/g. The surface morphology of carbon material in Fig. 9i obtained from the MCM-41 template resembles nanotubes without an ordered arrangement. The same result was also confirmed by SAXS analysis (data not shown). This observation indicates that MCM-41 silica was unsuitable as a template for OMC synthesis (Ryoo et al. 2001). Figure 9j–l shows the carbon samples obtained using a MCM-48 as template. The carbon formed without the addition of sulfuric acid reveals a corrugated tissue (Fig. 9j). The carbon deposition on the sample surface indicated that sucrose does not easily penetrate the silica pores. Figure 9k, l shows the CMK-1 carbons obtained using sucrose and glucose as carbon precursors by adding a sulfuric acid catalyst. The appearance of the two carbon materials is similar to MCM-48 silica (Kaneda et al. 2002). As expected from the results of SAXS analysis in Fig. 6, H2SO4 pre-treatment successfully converted sucrose and glucose to ordered mesoporous carbons.

FE-SEM images of samples: a–f mesoporous silica samples synthesized at various gelation pH values of 1, 3, 5, 7, 9, and 11, respectively; g mesoporous silica with lamellar structure; h carbon sample obtained from direct carbonization of as-synthesized MCM-48 composite; i carbon sample obtained using a MCM-41 as template; j carbon sample obtained without the addition of sulfuric acid; and k–l carbon samples obtained using sucrose and glucose as carbon precursors, respectively
Figure 10a shows the typical PRA powder after burning the packaging resin waste in air. The size of the particles was approximately 60-nm long and 10-nm wide. Figure 10b is a low-resolution TEM image of MCM-48 particles, showing a well-defined edge and a rectangular particle shape. Figure 10c, d shows the high-resolution TEM images of MCM-48 particles. The sample shows a regular array of mesopores along the (111) direction (Fig. 10c), and a uniform pore structure along the (100) direction (Fig. 10d). These images indicate that the cubic mesostructure of MCM-48 is well developed, which is in agreement with the results of SAXS (Fig. 1d). Wang et al. (Wang et al. 2005) observed similar morphologies. Typical HR-TEM images of OMC samples are shown in Fig. 10e, f. Figure 10e shows a pattern of parallel stripes with black and white in sharp contrast. The carbon materials consisted of highly ordered mesopores. Figure 10f shows rectangular edges of the carbon particles with a uniform pore distribution. No carbon deposition is apparent on the external surface of OMC, indicating that the amount of sucrose was just sufficient to fill the internal cellular pores of MCM-48.

High-resolution TEM images of samples: a PRA, b particle morphology of MCM-48, c, d MCM-48 synthesized at pH 11, e, f CMK-1 carbon
Conclusion
MCM-48 mesoporous molecular sieves free of Na impurities were synthesized using cheap, recycled silica from electronic packaging resin ash. The structure of silica samples achieved using different pH media, hydrothermal temperatures, and ratios of surfactant changed continuously from hexagonal to cubic to lamellar. Results confirm that ordered CMK-1 carbon can be successfully synthesized from the MCM-48 template using sucrose as a carbon precursor. The conversion of packaging resin ash into MCM-48 offers a better alternative for recycling and utilization of e-waste. Both MCM-48 and CMK-1 materials reveal a high surface area and uniform pore size, rendering them suitable for the development of advanced materials in energy and green application.
References
Bhagiyalakshmi M, Yun LJ, Anuradha R, Jang HT (2010a) Utilization of rice husk ash as silica source for the synthesis of mesoporous silicas and their application to CO2 adsorption through TREN/TEPA grafting. J Hazard Mater 175:928–938
Bhagiyalakshmi M, Lee JY, Jang HT (2010b) Synthesis of mesoporous magnesium oxide: Its application to CO2 chemisorption. Int J Greenh Gas Control 4:51–56
Brinker CJ, Scherer GW (1990) Applications. In: Sol–gel science, the physics and chemistry of sol–gel processing. Academic Press, Inc., San Diego
Chandrasekar G, Ahn WS (2008) Synthesis of cubic mesoporous silica and carbon using fly ash. J Non-Cryst Solids 354:4027–4030
Chen D, Bi X, Liu M, Huang B, Sheng G, Fu J (2011) Phase partitioning, concentration variation and risk assessment of polybrominated diphenyl ethers (PBDEs) in the atmosphere of an e-waste recycling site. Chemosphere 82:1246–1252
Cui J, Zhang L (2008) Metallurgical recovery of metals from electronic waste: a review. J Hazard Mater 158:228–256
Dai M, Song L, LaBelle JT, Vogt BD (2011) Ordered mesoporous carbon composite films containing cobalt oxide and vanadia for electrochemical applications. Chem Mater 23:2869–2878
Doyle A, Hodnett BK (2003) Stability of MCM-48 in aqueous solution as a function of pH. Microporous Mesoporous Mater 63:53–57
Doyle AM, Ahmed E, Hodnett BK (2006) The evolution of phases during the synthesis of the organically modified catalyst support MCM-48. Catal Today 116:50–55
Han S, Xu J, Hou W, Yu X, Wang Y (2004) Synthesis of high-quality MCM-48 mesoporous silica using gemini surfactant dimethylene-1,2-bis(dodecyldimethylammonium bromide). J Phys Chem B 108:15043–15048
Hao GP, Li WC, Wang S, Zhang S, Lu AH (2010) Tubular structured ordered mesoporous carbon as an efficient sorbent for the removal of dyes from aqueous solutions. Carbon 48:3330–3339
Hussain M, Ihm SK (2009) Synthesis, characterization, and hydrodesulfurization activity of new mesoporous carbon supported transition metal sulfide catalysts. Ind Eng Chem Res 48:698–707
Jang HT, Park YK, Ko YS, Lee JY, Margandan B (2009) Highly siliceous MCM-48 from rice husk ash for CO2 adsorption. Int J Greenh Gas Control 3:545–549
Joo SH, Jun S, Ryoo R (2001) Synthesis of ordered mesoporous carbon molecular sieves CMK-1. Microporous Mesoporous Mater 44–45:153–158
Kaneda M, Tsubakiyama T, Carlsson A, Sakamoto Y, Ohsuna T, Terasaki O, Joo SH, Ryoo R (2002) Structural study of mesoporous MCM-48 and carbon networks synthesized in the spaces of MCM-48 by electron crystallography. J Phys Chem B 106:1256–1266
Kim TW, Chung PW, Slowing II, Tsunoda M, Yeung ES, Lin VSY (2008) Structurally ordered mesoporous carbon nanoparticles as transmembrane delivery vehicle in human cancer cells. Nano Lett 8:3724–3727
Kim H, Karkamkar A, Autrey T, Chupas P, Proffen T (2009) Determination of structure and phase transition of light element nanocomposites in mesoporous silica: case study of NH3BH3 in MCM-41. J Am Chem Soc 131:13749–13755
Kim HJ, Han B, Kim YJ, Yoa SJ (2010) Characteristics of an electrostatic precipitator for submicron particles using non-metallic electrodes and collection plates. J Aerosol Sci 41:987–997
Kruk M, Jaroniec M, Ryoo R, Joo SH (2000) Characterization of MCM-48 silicas with tailored pore sizes synthesized via a highly efficient procedure. Chem Mater 12:1414–1421
Lelong G, Bhattacharyya S, Kline S, Cacciaguerra T, Gonzalez MA, Saboungi ML (2008) Effect of surfactant concentration on the morphology and texture of MCM-41 materials. J Phys Chem C 112:10674–10680
Liou TH (2003) Pyrolysis kinetics of electronic packaging material in a nitrogen atmosphere. J Hazard Mater 103:107–123
Liou TH (2004) Kinetics study of thermal decomposition of electronic packaging material. Chem Eng J 98:39–51
Liou TH (2010) Development of mesoporous structure and high adsorption capacity of biomass-based activated carbon by phosphoric acid and zinc chloride activation. Chem Eng J 158:129–142
Liou TH (2011) A green route to preparation of MCM-41 silicas with well-ordered mesostructure controlled in acidic and alkaline environments. Chem Eng J 171:1458–1468
Liou TH, Wu SJ (2009) Characteristics of microporous/mesoporous carbons prepared from rice husk under base- and acid-treated conditions. J Hazard Mater 171:693–703
Liou TH, Wu SJ (2010) Kinetics study and characteristics of silica nano-particles produced from biomass-based material. Ind Eng Chem Res 49:8379–8387
Liou TH, Yang CC (2011) Synthesis and surface characteristics of nanosilica produced from alkali-extracted rice husk ash. Mater Sci Eng B-Adv 176:521–529
Liu X, Du Y, Guo Z, Gunasekaran S, Ching CB, Chen Y, Leong SSJ, Yang Y (2009) Monodispersed MCM-41 large particles by modified pseudomorphic transformation: direct diamine functionalization and application in protein bioseparation. Microporous Mesoporous Mater 122:114–120
Liu A, Han S, Che H, Hua L (2010) Fluorescent hybrid with electron acceptor methylene viologen units inside the pore walls of mesoporous MCM-48 silica. Langmuir 26:3555–3561
Peña ML, Kan Q, Corma A, Rey F (2001) Synthesis of cubic mesoporous MCM-48 materials from the system SiO2:CTAOH/Br:H2O. Microporous Mesoporous Mater 44–45:9–16
Peng X, Cao D, Wang W (2008) Heterogeneity characterization of ordered mesoporous carbon adsorbent CMK-1 for methane and hydrogen storage: GCMC simulation and comparison with experiment. J Phys Chem C 112:13024–13036
Petitto C, Galarneau A, Driole MF, Chiche B, Alonso B, Renzo FD, Fajula F (2005) Synthesis of discrete micrometer-sized spherical particles of MCM-48. Chem Mater 17:2120–2130
Prasanth KP, Raj MC, Bajaj HC, Kim TH, Jasra RV (2010) Hydrogen sorption in transition metal modified mesoporous materials. Int J Hydrogen Energy 35:2351–2360
Ryoo R, Joo SH, Kim JM (1999) Energetically favored formation of MCM-48 from cationic—neutral surfactant mixtures. J Phys Chem B 103:7435–7440
Ryoo R, Joo SH, Kruk M, Jaroniec M (2001) Ordered mesoporous carbons. Adv Mater 13:677–681
Salgado JRC, Alcaide F, Alvarez G, Calvillo L, Lazaro MJ, Pastor E (2010) Pt–Ru electrocatalysts supported on ordered mesoporous carbon for direct methanol fuel cell. J Power Sources 195:4022–4029
Samadi-Maybodi A, Teymouri M, Vahid A, Miranbeigi A (2011) In situ incorporation of nickel nanoparticles into the mesopores of MCM-41 by manipulation of solvent-solute interaction and its activity toward adsorptive desulfurization of gas oil. J Hazard Mater 192:1667–1674
Sayari A (2000) Novel synthesis of high-quality MCM-48 silica. J Am Chem Soc 122:6504–6505
Shih LH (2001) Reverse logistics system planning for recycling electrical appliances and computers in Taiwa. Resour Conserv Recycl 32:55–72
Sing KSW, Everett DH, Haul RAW, Moscou L, Pierotti RA, Rouquerol J, Siemieniewska T (1985) Reporting physisorption data for gas/solid systems with specific reference to the determination of surface area and porosity. Pure Appl Chem 57:603–619
Wang LZ, Shi JL, Yu J, Zhang WH, Yan DS (2000) Temperature control in the synthesis of cubic mesoporous silica materials. Mater Lett 45:273–278
Wang L, Shao Y, Zhang J (2005) Short-time formation of well-ordered cubic mesoporous MCM-48 molecular sieve with the aid of fluoride ions. Mater Lett 59:3604–3607
Wei FY, Liu ZW, Lu J, Liu ZT (2010) Synthesis of mesoporous MCM-48 using fumed silica and mixed surfactants. Microporous Mesoporous Mater 131:224–229
Ying F, Wang S, Au CT, Lai SY (2011) Highly active and stable mesoporous Au/CeO2 catalysts prepared from MCM-48 hard-template. Microporous Mesoporous Mater 142:308–315
Zhao W, Hao Z, Hu C (2005) Synthesis of MCM-48 with a high thermal and hydro-thermal stability. Mater Res Bull 40:1775–1780
Zhao D, Budhi S, Rodriguez A, Koodali RT (2010a) Rapid and facile synthesis of Ti-MCM-48 mesoporous material and the photocatalytic performance for hydrogen evolution. Int J Hydrogen Energy 35:5276–5283
Zhao W, Li Q, Wang L, Chu J, Qu J, Li S, Qi T (2010b) Synthesis of high quality MCM-48 with binary cationic—nonionic surfactants. Langmuir 26:6982–6988
Acknowledgments
The author expresses thanks to the National Science Council of Taiwan for its financial support under Project No. NSC 96-2221-E-131-001.
Author information
Authors and Affiliations
Corresponding author
Additional information
Special Issue Editors: Mamadou Diallo, Neil Fromer, Myung S. Jhon
This article is part of the Topical Collection on Nanotechnology for Sustainable Development
Rights and permissions
About this article
Cite this article
Liou, TH. Recovery of silica from electronic waste for the synthesis of cubic MCM-48 and its application in preparing ordered mesoporous carbon molecular sieves using a green approach. J Nanopart Res 14, 869 (2012). https://doi.org/10.1007/s11051-012-0869-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-012-0869-6