
Abstract
Fullerenes are sparingly soluble in many solvents. The dependence of fullerene’s solubility on molecular structure of the solvent must be understood in order to manage efficiently this class of compounds. To find such dependency ab initio quantum-chemical calculations in combination with quantitative structure–property relationship (QSPR) tool were used to model the solubility of fullerene C60 in 122 organic solvents. A genetic algorithm and multiple regression analysis (GA-MLRA) were applied to generate correlation models. The best performance is accomplished by the four-variable MLRA model with prediction coefficient r 2test = 0.903. This study reveals a correlation of highest occupied molecular orbital energy (HOMO), certain heteroatom fragments, and geometrical parameters with solubility. Several other important parameters of solvents that affect the C60 solubility have been also evaluated by the QSPR analysis. The employed GA-MLRA approach enhanced by application of quantum-chemical calculations yields reliable results, allowing one to build simple, interpretable models that can be used for predictions of C60 solubility in various organic solvents.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
It is well known that the fullerenes are sparingly soluble in various solvents (Ruoff et al. 1993). This phenomenon has crucial consequences for both the basic studies on fullerenes and their industrial applications. The dependence of fullerene’s solubility on both molecular structure of the solvent and temperature must be understood in order to efficiently separate different members of the fullerene family from each other and from their precursors or derivatives (Korobov and Smith 2000). Though experimental data on fullerene solubility is available, there is still no reliable theory to explain (or predict) absolute values of fullerene solubility and variations in solubility when changing the solvent or using different fullerenes. This serious limitation is one of the reasons why separation of fullerenes on a large scale is still rather difficult without the aid of chromatography. Moreover, the data on solubility of the fullerene C60 in organic solvents could help to develop its various applications in chemistry and technology.
At present, the only approach allowing construction of predictive model of fullerene C60 solubility in different organic solvents is quantitative structure–property relationship (QSPR) based on molecular descriptors (i.e., parameters) calculated with molecular graphs of the solvents (Estrada and Gutman 1996; Simon 1987; Balaban 1983). The first attempt to explain the trends in C60 solubility was carried out by Sivaraman et al. (1992) who found a correlation between experimental fullerene’s solubility and calculated solubility parameter of solvent by studying fullerene’s solubility for 15 solvents. The same group also published another study devoted to fullerene solubility. For a series of solvents they plotted the fullerene solubility versus the Hildebrand solubility parameter (Sivaraman et al. 1994). In another study, Ruoff et al. (1993) studied the C60 solubility in 47 solvents. They examined the dependence of solubility on the polarizability, polarity, molecular size, and cohesive energy density (energy difference between solid phase and free atoms system) of the solvents. However, they found no distinctive parameter that universally explains the solubility of C60 and no predictive model was built in their study. Smith et al. (1996) employed the theoretical linear solvation energy approach with solubility dataset of 101 organic solvents. The correlation coefficient was r 2 = 0.67 and statistically significant descriptors that were revealed in their study are molar volume, Lewis acidity, Lewis basicity, and electrostatic basicity. Thus, a good solvent for C60 should have a large molar volume and also be both a good Lewis acid and a good Lewis base. However, it should possess minimal polarity of the regions of negative charge. Though it is an interesting idea, the drawback of this study is too low correlation coefficient of the modeled relationship. In addition, a study has been published where authors aimed to apply molecular dynamics approach to model the solubility of fullerene in two solvents, water and carbon disulfide (Vanin et al. 2008). However, this approach is extremely time-consuming to apply it for large dataset of solvents and therefore not desirable to use in QSPR. There were also other articles regarding the solubility of C60 in different solvents (Marcus 1997, Marcus et al. 2001; Abraham et al. 2000; Hansen and Smith 2004; Kiss et al. 2000). The recent study in this field was published by Liu et al. (2005). The authors used the biggest to date dataset of 128 solvents. They built a model using least-squares support vector machine (LSSVM) method and the obtained correlation coefficients were r 2 = 0.761 for the whole set and r 2 = 0.861 for the 122 compounds (six compounds, i.e., outliers, removed). The correlation coefficients for the splitted dataset to training (92 compounds) and test (30 compounds) sets were 0.910 and 0.908, respectively. The authors used CODESSA software (Katritzky et al. 1994, 1995) to generate a set of descriptors and semiempirical quantum–mechanical approach to calculate quantum-chemical parameters for the considered solvents. They showed that such descriptors as Randic index (order 3), relative molecular weight, HOMO-1 energy (highest occupied molecular orbital), relative negative charge, average bonding information content, and average one-electron reactivity index for a C atom are involved in correlation model. In our recent study (Toropov et al. 2007a) devoted to solubility of C60 the QSPR model based on SMILES (Simplified Molecular Input line Entry System) notations and optimal descriptors was developed. This model allows to achieve correlation coefficient for the training set of r 2 = 0.861 and for the external set of r 2test = 0.890. The approach implemented in the current study differs considerably from the previous studies because in the earlier investigations only a very simple linear model with one complex parameter (correlation weight based on SMILES notation) was used that provided a good predictive ability. In addition, recently the authors of this study in collaboration with the experimental biology group published the study where they aimed to choose a better solvent for C60 fullerene in biological systems, based on cytotoxicity tests (Cook et al. 2010).
As it was pointed out above, there are many different approaches to calculate and predict C60 solubility in organic solvents. Some of them are fully mechanistic (Marcus 1997, Marcus et al. 2001; Abraham et al. 2000; Hansen and Smith 2004; Stukalin et al. 2003), developed from the thermodynamical point of view; others are statistically based, with good correlation coefficients, but not transparent and complicated in interpretation (Kiss et al. 2000; Liu et al. 2005; Toropov et al. 2007a, b, 2008, 2009). In this study, we aimed to find simple, transparent relationship and computationally fast approach, possibly mechanistically interpretable, to predict the solubility of C60 in various solvents. In addition, the study intends to estimate predictive potential of the topological descriptors and quantum-chemical parameters obtained by high level ab initio calculations in QSPR modeling of the fullerene C60 solubility in organic solvents.
Methods
Data preparation
The splitting of the experimental data into training and test sets for solubility of C60 in 122 different solvents was taken from ref (Liu et al. 2005), but originally these data were published previously in review paper (Beck and Mandi 1997). The solvents’ data are listed in Table S1 (see Supplementary material) and in Table 2. The solubilities are not given in weight units (e.g., mg/mL), but in terms of logarithmic values of molar fractions (log S) because the log S values correspond to the free energy changes in the solvation process. The data splitting into training and test sets was carried out according to refs. (Liu et al. 2005; Toropov et al. 2007c), i.e., to training set of 92 solvents and test set of 30 solvents, where it was splitted randomly.
Quantum-chemical calculations and statistical GA-MLRA approach
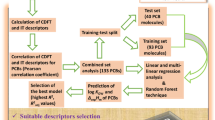
To find proper parameters that are responsible for C60 solubility in organic solvents we used two approaches: (1) structurally based additive descriptors calculation, and (2) quantum-chemically calculated descriptors. The set of descriptors obtained by the first approach represents the set of constitutional, topological, and molecular descriptors that were calculated by the DRAGON software (Todeschini and Consonni 2003). A set of 1,060 molecular descriptors of different types was used to describe the chemical diversity of the considered solvent compounds. The descriptor’s typology involves: (a) constitutional (atom and group fragments), (b) functional groups, (c) atom-centered fragments, (d) empirical, (e) topological, (f) walk counts, (g) various autocorrelations from the molecular graph, (h) Randic molecular profiles from the geometry matrix, (i) geometrical, (j) WHIM, and (k) GETAWAY descriptors, and various indicator descriptors. The meaning of these molecular descriptors and the calculation procedures are summarized elsewhere (Todeschini and Consonni 2000).
Considering the importance of the electronical molecular properties for QSAR/QSPR, the additional descriptors were calculated by the second approach—the quantum-chemical descriptors (Hehre et al. 1986; Becke 1993). Quantum-chemical descriptors have been calculated using Gaussian 03 software (Frisch et al. 2004) by Density Functional Theory (DFT) methodology. The global minimum-energy conformation was identified for each molecule. A combination of Becke’s three-parameter adiabatic connection exchange functional with Los Alamos National Lab effective core potential with double-zeta basis set for valence electrons (B3LYP/LANL2DZ) was employed in order to obtain reliable energetics and accurate data on electronic properties of the considered molecules. Some of considered solvents contain Br and I atoms and therefore LANL2DZ basis set has been applied. The LANL2DZ basis set has been developed for predictions of heavy atoms, beyond the third row (Foresman and Frisch 2000). Overall, different kinds of quantum-chemical descriptors were used based on performed calculations, including dipole moments (total dipole moment, X, Y, and Z components); orbital energies, EHOMO, ELUMO; HLgap (gap between EHOMO and ELUMO), and finally, heats of formation and ionization potentials.
The correlation coefficients for all pairs of descriptor variables used in the applied models were evaluated in order to identify highly correlated descriptors and to avoid redundancy in the data set. Any type of redundancy might lead to an overexploitation of a chemical property in the explanation of the dependent variable. Hence, some highly correlated and constant descriptors (cross-correlation r 2 > 0.9) were removed from the further consideration. Furthermore, at the process of each model building (i.e., inside of each final model), the descriptors with cross-correlation coefficient larger than 0.6 are avoided.
The correlation between solubility and structural properties was obtained by using a variable selection Genetic Algorithm (GA) and MLRA methods. GA has been applied in recent years as a powerful tool to address many problems in drug design (Davis 1991; Devillers 1996). We applied GA to select from the set of all calculated descriptors only the best combinations of those the most relevant for obtaining models with the highest predictive power of solubility. Overall, the combined GA-MLRA technique was utilized to select the appropriate descriptors and to generate different QSPR models. The GA technique started with a population of 30 random models and was carried out up to 5500 iterations for evolution, mutation—35%, fitness/scoring function—correlation coefficient (r). For GA analysis and the derivation of the QSPR models, the BuildQSAR program (de Oliveira and Gaudio 2001) was used. A final set of QSARs was validated by applying the “leave-one-out” technique with its predicting ability being evaluated and confirmed by cross-validation coefficient q 2.
Results
Statistical characteristics of the best one-, two-, three-, four- and five-variable models are shown in Table 1. As a rule of thumb, for providing reliable results we calculated different models by increasing number of involved descriptors until the model with a higher number of descriptors gives lower prediction coefficient (because of overfitting) for the test set, in comparing to the previous model (Figure S1 (Supplementary material)).
The solvents’ names and CAS numbers for all used compounds and also descriptors’ names, which have been involved in all best models, are showed in the Table S1 of Supplementary material. One can see from the data in Table 1 that the four-variable model (Eq. 4) yields the best predictive potential for the solubility of C60 in organic solvents. While constructing the models, great care was taken in order to avoid inclusion of highly collinear descriptors. All collinear descriptors were eliminated from the further consideration.
One-variable model can be described as follows:
Two-variable model:
Three-variable model:
Four-variable model:
Five-variable model:
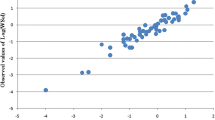
Graphical representation of the experimental and predicted solubility values according to the best model (Eq. 4) for the training and test sets are shown in Fig. 1. Experimental and predicted values of log S for Eq. 4 are displayed in Table 2.

Plot of experimental versus calculated fullerene C60 solubility for the training and test sets
As it can be seen, all models include mostly well explainable and transparent descriptors, including quantum-chemical parameters.
The first four models include topological descriptor X1sol, which represents solvation connectivity index (chi-1) that encodes the solvation property of the compound (Todeschini and Consonni 2003). This molecular descriptor is defined in order to model solvation entropy and dispersion interactions in solution. The descriptor relates the characteristic dimension of the molecule to the atomic parameters (quantum number, bond indexes, etc.). The bidimensional descriptor X1sol was proposed in 1991 by the group of Zefirov and Palyulin (Antipin et al. 1991) in order to treat the enthalpies of non-specific solvation. For instance, as it was described in this study (Duchowicz et al. 2008), the solvation enthalpy of propane (CH3CH2CH3) and dimethyl-mercury (CH3HgCH3) differs enormously, but both of these molecules are represented by the same hydrogen depleted graph, and, hence, they have the identical topological indexes, which do not take into account the atom types. The solvation index was created exactly to differentiate such cases, providing the general formula (see below) when calculated for hydrogen- and fluorine-depleted molecular graphs.
The descriptor is defined by the following equations. If the characteristic dimensions of the molecules by atomic parameters are taken into account, they are defined as:
where L a is the principal quantum number (2 for C, N, O atoms, 3 for Si, S, Cl, etc.) of the ath atom in the kth subgraphs; δ a represents the corresponding vertex degree; k is the total number of mth order subgraphs and n is the number of vertices in the subgraph. The normalization factor 1/(2m+1) is defined in such a way that the indexes Xm and Xmsol for compounds, which contain only second-row atoms do coincide. The 1st order solvation connectivity index is
where b runs over all the B bonds; L i and L j are the principal quantum numbers of the two vertices related to the considered bond. The positive coefficient of X1sol indicates that an increase in the descriptor value results in an increase in solubility of C60 in the considered solvent (see Fig. 3). The J3D descriptor represents 3D-Balaban index, the geometrical descriptor (Todeschini and Consonni 2003). The Balaban index describes the distance connectivity of the molecule, which is the average distance sum connectivity. J3D contributes negatively to the solubility of C60 in a solvent. HOMO descriptor represents a quantum chemical descriptor, the energy of the highest occupied molecular orbital, obtained by the B3LYP/LANL2DZ level calculations using the DFT approach. This descriptor describes the nucleophilic properties of solvent. According to Eq. 3 the higher energy of the HOMO of solvent results in the higher solubility of C60. The next similar descriptor from quantum-chemical calculations is the HOMO–LUMO gap, which also shows a good correlation with the C60 solubility (see Fig. 3). The TI2 descriptor is topological descriptor, second Mohar index TI2. The Mohar index is derived from Laplacian matrix (Todeschini and Consonni 2003; Mohar 1989), a distance matrix. As it can be seen from the Eq. 4, the higher value of TI2 descriptor results in the lower solubility of C60 in this solvent. Another important descriptor selected by genetic algorithm is FDI descriptor, which is geometrical descriptor representing a folding degree index. The FDI descriptor is defined as the largest eigenvalue obtained by the diagonalization of the distance/distance matrix, and then normalized and divided by the number of atoms (Todeschini and Consonni 2003; Randic et al. 1994; Randic and Krilov 1999). The values of the descriptor are in range 0 ≤ FDI ≤ 1. This descriptor converges to one for linear molecules (of infinite length) and decreases in accord with the folding degree of the molecule. The FDI descriptor can be used as indicator of the degree of departure of a molecule from a strict linearity. FDI contributes positively to the log S value of C60 solubility. It indicates that the more linear (less foldings) molecule of the solvent is, the better is solubility of C60 in this solvent. The H-052 descriptor is among atom-centered fragments, describing H (hydrogen) attached to C(sp3) with 1X (heteroatom) attached to the next C (Todeschini and Consonni 2003). The higher value of this descriptor relates to the lower solubility of C60. The nHAcc descriptor represents a number of acceptor atoms for H bonds (N, O, F, etc.). This descriptor is among the functional group descriptors. X3 descriptor represents connectivity index chi-3, and this is a topological descriptor (Todeschini and Consonni 2003). And AMW, a constitutional descriptor that describes an average molecular weight. This descriptor shows a positive correlation with the solubility of C60.
Discussion
In the recently published book on fullerenes (Korobov and Smith 2000), a statement was given that there is no single parameter to correlate C60 solubility. Our comprehensive study reveals that one X1sol descriptor provides good, and quite satisfactory correlation for both sets—training and test set (r 2 = 0.641 and r 2test = 0.502). The ability of this descriptor to encode several important for solubility characteristics in one parameter makes it a good predictor for C60 solubility. Of course, the predictive ability of this descriptor is still not enough for complete estimation, but it can be used for fast preliminary evaluation of fullerene solubility. One might note that X1sol contributes to all four best models and that the contribution of this descriptor to each model is significant. As it can be seen from the Fig. 2, the X1sol descriptor describes C60 solubility very well for each compound. There are even no evident outliers among the studied solvents (except of tetradecane (1) in the test set), although all points are distributed slightly sparse along a trend line. The presence of tetradecane (1) as outlier is caused by high calculated value of X1sol, i.e., according to the molecular structure the tetradecane should have better solubility properties than actually observed. We believe that because of the linear structure of the tetradecane molecule some other factors are predominant for C60 solubility in this particular solvent. Interestingly, also the other descriptor FDI confirmed some prediction uncertainness for linear molecules like tetradecane, and this is discussed later in the text. Considering that this is only one-parameter model, one may conclude that the proposed model provides a good result, r 2test = 0.502. The descriptor basically encodes the presence of heteroatoms that mainly determine the solvent properties, as well as number and connectivity between subgraphs that include these heteroatoms. Such subgraphs determine the strength and pattern of the lattice that is built by the molecules of solvent, and this lattice, most likely, determines the C60 dissolution rate in the solvent. This conclusion is in accordance with explanation from the recent study (Herbst et al. 2005). The authors of this study underlined that H-bonding character of the solvent is an obvious discriminating factor and C60 has a lower solubility in solvents that organize themselves through polar or H-bond interactions, because the dissolution of C60 would result in the disruption of the solvent structure since the nonpolar solute does not participate in H-bonds. The presence of nHAcc descriptor in the model 5 also confirms importance of H bonds.

Plot of solubility of C60 versus X1sol descriptor for both sets
The two- and three-variable models (Eqs. 2, 3) include J3D descriptor that encodes heteroatoms' content and bond multiplicity. This descriptor improves the performance of two-variable model greatly, from r 2 = 0.6 for one-variable model to almost 0.8 for the two-variable one. The J3D descriptor is not cross-correlated with X1sol descriptor. It encodes different features and only informationally supplements the X1sol descriptor as well as amplifies the model performance. The J3D descriptor indicates the importance of heteroatoms' content in solvent structure, because the presence of heteroatoms dramatically changes the solubility property of the compound. Although both X1sol and J3D descriptors partially encode the presence of heteroatoms in the molecule, they work independently and only improve the performance of each other.
Looking for the best performance model that works better for the prediction of the test set values we generated four- and five-variable models. We finally establish that four-variable model has the best performance, its predictive r 2 value for the test set is 0.903, including all compounds (Table 1, Fig. 1). The model 5 (Eq. 5) shows much worse performance for the test set (most) probably because of overfitting.
The other two worthy of note descriptors are HOMO energy and HOMO-LUMO gap (HLgap). The Eqs. 3 and 5 illustrate importance of these parameters (HOMO and HLgap). There is a correlation between them, i.e., they encode mainly similar information. Such a correlation between these two parameters can be explained by main contribution of HOMO energy parameter to HOMO–LUMO gap. After closer analysis of the HOMO–LUMO gap parameter one can see that there are three different types of solvents. They markedly differ from each other—alkanes (a), alcohols (b) and other compounds (c), which contain heteroatom(s) (see Fig. 3). This situation is observed in both sets (training and test), Fig. 3. It can be seen that compounds of the three groups are energetically very different and trends of “HOMO–LUMO gap versus the Solubility of C60” relationships are different for each group. It means that using only HOMO–LUMO gap parameter for each group of these solvents separately one can build more precise models for different solvent groups. However, our aim was to build universal C60 solubility model that can describe as many various compounds as possible.

Plot of solubility C60 data versus HOMO–LUMO gap for both sets: (a) alkanes, (b) alcohols, (c) other compounds (heteroatom-containing)
Rather interesting descriptor, involved in Eq. 4 is H-052, the number of hydrogens at the considered molecular fragment. One concludes that large number of substituents at the carbon atoms that are next to carbon atom with heteroatom results in lower solubility. But this statement is only exact for solvents that do not have full substitution at the neighbor carbon atoms, i.e., where there are still 1–4 hydrogen atoms. Interestingly, in case of unsubstituted neighbor carbon atoms the solubility in such solvents became lower (H-052 = 6). Furthermore, the solvents with fully substituted carbon atoms (no hydrogens at the neighbors around the carbon atom with heteroatom) are the best solvents for C60, and their behavior differs from the others, hydrogen-containing solvents.
Another important descriptor for C60 solubility is the FDI descriptor. According to the plot of FDI descriptor and solubility data one can see that there is a positive correlation—the higher FDI value, the higher solubility is. But this relationship works fine only for nonlinear molecules (folded molecules), one can see an apparent trend for such molecules (Fig. 4). In the case of linear molecules (FDI = 1, i.e., no foldings) the situation is not clear. Most likely, the other factors (which were mentioned before) also play an important role in solubility for both types of solvents, but in the case of molecules that have molecular structures close to linear the other factors are predominant (such factors as molecular weight AMW, HOMO energy, HOMO–LUMO gap, number and type of heteroatoms, etc.).

Plot of solubility of C60 versus FDI descriptor for both sets
As it was showed above, all descriptors that are included in the models are quite simple and transparent. They can easily be calculated using available software, and by using the developed models the predictive value of C60 solubility in a particular solvent can be calculated. This is one of the advantages of the models obtained in this study, comparing to Liu et al. (2005) investigation, where models are not represented and therefore can not be reproduced to predict solubility for new solvents.
Overall, among all models generated the four-variable model is a good candidate for further use for C60 solubility predictions. It has a good predictive power, transparent descriptors and includes only easy-to-calculate descriptors. The contribution and structural influence of each descriptor involved in four-variable model is discussed above.
Conclusions
In this study we have compared the performances of one-, two-, three- four- and five-variable models constructed by GA-MLRA approach for prediction of solubility of C60. The four-variable model provides the best predictive ability (r 2test = 0.903), while the other models give only satisfactory prediction values. For one-, two-, and three-variable models it can be explained by insufficient information providing by the small number of descriptors in the model, and for five-variable model it is due to its overfitting that leads to bad prediction.
The study reveals a correlation of HOMO energy (HOMO–LUMO gap), heteroatom fragments and geometrical parameters with solubility. Although the X1sol descriptor alone displayed the statistically not so significant correlation, it still provides a main contribution to solubility. The presence of quantum-chemical descriptor—HOMO energy confirmed the importance of nucleophilic properties of solvents for solubility of C60. Higher HOMO value for solvent results in the higher solubility of C60. Also, various distribution behavior of HOMO–LUMO gap energies for different types of solvents have been concluded. Another descriptor, which encodes number of hydrogens at certain fragments (H-052), indicates that the solvents with fully substituted carbon atoms (no hydrogens at the neighbors around the carbon atom with heteroatom) are the best solvents for C60, and their behavior differs from the others, hydrogen-containing solvents. Surprisingly, the folding descriptor FDI showed certain correlation with solubility, asserting that the closer structure of solvent to linearity the higher solubility of C60 in this solvent is. Several other important parameters of solvents that affect C60 solubility also have been discussed based on the QSPR analysis. They lead to better understanding of the solvent structure required for improving of the fullerene solubility.
This study demonstrates that an application of the GA-MLRA technique in combination with quantum-chemical and topological descriptors yields reliable models. They are quite simple, interpretable, transparent, and comparable to the previously published results. The best performance is accomplished by the four-variable MLRA model with prediction coefficient r 2test = 0.903. An applied approach based on topological and quantum-chemical data provides the model for fullerene C60 solubility which is comparable with the model from the previous study (Liu et al. 2005) and model suggested in the recently published paper (Toropov et al. 2007c), in addition of having an advantage of being transparent and mechanistically interpretable. These conclusions allow us to believe that the constructed models can be used for future predictions of C60 solubility in various organic solvents (for industry and laboratory experiments) and that they provide basics for understanding of this phenomenon.
References
Abraham MH, Green CE, Acree WE (2000) Correlation and prediction of the solubility of Buckminsterfullerene in organic solvents; estimation of some physicochemical properties. J Chem Soc Perkin Trans 2:281–286
Antipin IS, Arslanov NA, Palyulin VA, Konovalov AI, Zefirov NS (1991) Solvation topological index. Topological description of dispersion interaction (in Russian). Dokl Akad Nauk SSSR 316:925–927 (Chem Abstr 115, 91390)
Balaban AT (1983) Topological indexes based on topological distances in molecular graphs. Pure Appl Chem 55:199–206
Beck MT, Mandi G (1997) Solubility of C60. Fuller Sci Technol 5:291–310
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652
Cook SM, Aker WG, Rasulev BF, Hwang H-M, Leszczynski J, Jenkins JJ, Shockley V (2010) Choosing safe dispersing media for C60 fullerenes by using cytotoxicity tests on the bacterium Escherichia coli. J Hazard Mater 176:367–373
Davis L (1991) Handbook of genetic algorithms. Van Nostrand Reinhold, New York, USA
de Oliveira DB, Gaudio AC (2001) BuildQSAR: a new computer program for QSAR analysis. Quant Struct Act Relat 19:599–601
Devillers J (1996) Genetic algorithms in molecular modeling. Academic Press Ltd, London
Duchowicz PR, Talevi A, Bruno-Blanch LE, Castro EA (2008) New QSPR study for the prediction of aqueous solubility of drug-like compounds. Bioorg Med Chem 16:7944–7955
Estrada E, Gutman I (1996) A topological index based on distances of edges of molecular graphs. J Chem Inf Comp Sci 36:850–853
Foresman JB, Frisch A (2000) Exploring chemistry with electronic structure methods. Gaussian, Inc., Pittsburgh
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR et al (2004) Gaussian 03, Revision C.02. Gaussian, Inc., Wallingford
Hansen CM, Smith AL (2004) Using Hansen solubility parameters to correlate solubility of C60 fullerene in organic solvents and in polymers. Carbon 42:1591–1597
Hehre WJ, Radom L, Schleyer P, Pople JA (1986) Ab initio molecular orbital theory. Wiley, New York
Herbst MH, Dias GHM, Magalhaes JG, Torres RB, Volpe PLO (2005) Enthalpy of solution of fullerene [60] in some aromatic solvents. J Mol Liq 118:9–13
Katritzky AR, Lobanov VS, Karelson M (1994) Comprehensive descriptors for structural and statistical analysis. Version 2.0. Semichem, Inc., Gainesville (reference manual)
Katritzky AR, Lobanov VS, Karelson M (1995) Chem Soc Rev 24:279–287
Kiss IZ, Mandi G, Beck MT (2000) Artificial neural network approach to predict the solubility of C60 in various solvents. J Phys Chem A 104:8081–8088
Korobov MV, Smith AL (2000) Solubility of the fullerenes. In: Kadish KM, Ruoff RS (eds) Fullerenes: chemistry, physics, and technology. John Wiley and Sons Inc, New York, pp 53–90
Liu H, Yao X, Zhang R, Liu M, Hu Z, Fan B (2005) Accurate quantitative structure-property relationship model to predict the solubility of C60 in various solvents based on a novel approach using a least-squares support vector machine. J Phys Chem B 109:20565–20571
Marcus Y (1997) Solubilities of buckminsterfullerene and sulfur hexafluoride in various solvents. J Phys Chem 101:8617–8623
Marcus Y, Smith AL, Korobov MV, Mirakyan NV, Avramenko NV, Stukalin EB (2001) Solubility of C60 fullerene. J Phys Chem B 105:2499–2506
Mohar B (1989) Laplacian matrices of graphs. In: Graovac A (ed) MATH/CHEM/COMP 1988. Studies in physical and theoretical chemistry, vol 63, Elsevier, Amsterdam, pp 1–8
Randic M, Krilov G (1999) On a characterization of the folding of proteins. Int J Quantum Chem 75:1017–1026
Randic M, Kleiner AF, De Alba LM (1994) Distance/distance matrices. J Chem Inf Comp Sci 34:277–286
Ruoff RS, Tse DS, Malhotra R, Lorents DC (1993) Solubility of fullerene (C60) in a variety of solvents. J Phys Chem 97:3379–3383
Simon J (1987) Molecular graphs as topological objects in space. J Comp Chem 8:718–726
Sivaraman N, Dhamodaran R, Kaliappan I, Srinivasan TG, Rao PRV, Mathews CK (1992) Solubility of C60 in organic solvents. J Org Chem 57:6077–6079
Sivaraman N, Dhamodaran R, Kaliappan I, Srinivasan TG, Rao PRV, Mathews CK (1994) Solubility of C60 and C70 in organic solvents. In: Kadish KM, Ruoff RS (eds) Recent advances in the chemistry and physics of fullerenes and related materials. The Electrochemical Society, Pennington, pp 156–165
Smith AL, Wilson LY, Famini GR (1996) A quantitative structure-property relationship study of C60 solubility. In: Recent advances in the chemistry and physics of fullerenes and related materials, vol 3. Proceedings of Electrochemical Society, Philadelphia, pp 53–62
Stukalin EB, Korobov MV, Avramenko NV (2003) Solvation free energies of the fullerenes C60 and C70 in the framework of polarizable continuum model. J Phys Chem B 107:9692–9700
Todeschini R, Consonni V (2000) Handbook of molecular descriptors. Wiley-VCH, Weinheim
Todeschini R, Consonni V (2003) DRAGON software for the calculation of molecular descriptors Version 3.0
Toropov AA, Leszczynska D, Leszczynski J (2007a) QSPR study on solubility of fullerene C60 in organic solvents using optimal descriptors calculated with SMILES. Chem Phys Lett 441:119–122
Toropov AA, Leszczynska D, Leszczynski J (2007b) Predicting water solubility and octanol water partition coefficient for carbon nanotubes based on the chiral vector. Comput Biol Chem 31:127–128
Toropov AA, Rasulev BF, Leszczynska D, Leszczynski J (2007c) Additive SMILES based optimal descriptors: QSPR modeling of fullerene C60 solubility in organic solvents. Chem Phys Lett 444:209–214
Toropov AA, Rasulev BF, Leszczynska D, Leszczynski J (2008) Multiplicative SMILES-based optimal descriptors: QSPR modeling of fullerene C60 solubility in organic solvents. Chem Phys Lett 457:332–336
Toropov AA, Toropova AP, Benfenati E, Leszczynska D, Leszczynski J (2009) Additive InChI-based optimal descriptors: QSPR modeling of fullerene C60 solubility in organic solvents. J Math Chem 46(4):1232–1251
Vanin AA, Piotrovskaya EM, Piotrovsky LB (2008) Investigation of fullerene solutions by molecular dynamics method. Fuller Nanotub Car N 16(5–6):555–562
Acknowledgments
The authors would like to thank for support the National Science Foundation for the “Interdisciplinary Center for Nanotoxicity” support—NSF HRD #0833178; for NSF EPSCoR Grant no. 362492-190200-01\NSFEPS-0903787 and Department of Defense through the U. S. Army Engineer Research and Development Center (Vicksburg, MS) for the grant “Development of Predictive Techniques for Modeling Properties of NanoMaterials Using New OSPR/QSAR Approach Based on Optimal NanoDescriptors”—Contract #W912HZ-06-C-0061. The authors are grateful to the Mississippi Center for Supercomputing Research (MCSR) for providing state-of-the-arts high performance computing facilities and excellent services for supporting this research.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Petrova, T., Rasulev, B.F., Toropov, A.A. et al. Improved model for fullerene C60 solubility in organic solvents based on quantum-chemical and topological descriptors. J Nanopart Res 13, 3235–3247 (2011). https://doi.org/10.1007/s11051-011-0238-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11051-011-0238-x