
Abstract
Given the complexity of the airway microbiota in the respiratory tract of cystic fibrosis (CF) patients, it seems crucial to compile the most exhaustive and exact list of the microbial communities inhabiting CF airways. The aim of the present study was to compare the bacterial and fungal diversity of sputa from adult CF patients during non-exacerbation period by culture-based and molecular methods, and ultra-deep-sequencing (UDS). Sputum samples from four CF patients were cultured and analysed by DNA extractions followed by terminal restriction fragment length polymorphism analysis through resolution of bacterial ribosomal gene (rDNA) fragments, and cloning plus sequencing of part of fungal rRNA genes. These approaches were compared with UDS method targeting 16S rDNA gene and the internal transcribed spacer (ITS) 2 region of rDNA. A total of 27 bacterial and 18 fungal genera were detected from the four patients. Five (18%) and 3 (16%) genera were detected by culture for bacteria and fungi, respectively, 9 (33%) and 3 (16%) by first generation sequencing (FGS) methods, and 26 (96%) and 18 (100%) by UDS. The mean number of genera detected by UDS per patient was statistically higher than by culture or FGS methods. Patients with severe airway disease as assessed by standard spirometry exhibited a reduced fungal and bacterial diversity. UDS approach evaluates more extensively the diversity of fungal and bacterial flora compared with cultures. However, it currently remains difficult to routinely use UDS mainly because of the lack of standardization, and the current cost of this method.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The average life expectancy of patients with cystic fibrosis (CF) has greatly increased over the past 50 years, and this is arguably attributed to the introduction of antibiotic therapy to treat bacterial lung infections as the most important factor [1,2,3]. Despite this progress, gradual loss of lung function remains the cause of mortality in 90% of CF patients [1, 4]. In terms of laboratory investigations, Pseudomonas aeruginosa and Staphylococcus aureus bacteria are deemed responsible for the majority of respiratory tract exacerbations in adults CF patients and are often detected by conventional culture method [5,6,7]. However, other opportunistic microorganisms, such as Aspergillus spp. or anaerobes, can also cause such exacerbations, yet their detection using the conventional methods alone is sometimes difficult [8, 9]. Even if fungi are part of the rare biosphere, they still exhibit the ability to cause severe exacerbations [9,10,11]. In microbiology laboratory practice, conventional culture-based methods are the most widely used to diagnose fungal and bacterial exacerbations. These methods focused mostly on individual organisms rather than the whole microbial population and are only semi-quantitative. On the contrary, culture-independent molecular approaches can identify the entire microbial population present in respiratory samples but are also semi-quantitative. Molecular methods based on first generation sequencing (FGS), such as Terminal restriction fragment length polymorphism (T-RFLP) profiling for bacteria [12, 13] or cloning and sequencing the internal transcribed spacer region (ITS) of ribosomal DNA (rDNA) after PCR amplification (ITS–PCR cloning) for fungi [14], have been proposed to assess the microbial diversity of CF respiratory samples, but were seldom implemented in microbiological laboratories practice, mainly because they are time-consuming. More recently, ultra-deep sequencing (UDS) methods have been used to explore the airway microbiome in CF patients [6, 15, 16], but only a handful of studies looked at the presence of fungi and bacteria together [17,18,19,20]. However, the application range of this method in routine microbiology analyses of CF respiratory specimens remains to be defined.
The aim of the present study was to examine the bacterial and fungal communities, in terms of relative abundance and diversity, in four sputum samples from four adult CF patients during the non-exacerbation period using three methods: microbial cultures, molecular methods based on FGS, and UDS method which targets variable regions of bacterial 16S rDNA and the ITS2 region of fungal rDNA. We demonstrated the concordance of results obtained from culture and sequencing methods, highlighted comparison points for the most prevalent genera, and then we discussed the advantages and disadvantages of each method, in particular in microbiological laboratory investigations.
Materials and Methods
Patients and Samples
Patients with well-documented diagnosis of CF (homozygous or heterozygous for the F508del mutation) were eligible for the study if clinically stable (i.e. if they had no exacerbations over the entire year preceding the follow-up). A staff physician collected clinical data including spirometry, therapeutic, radiological and biological data at each medical visit. An ad hoc Ethics Committee (Comité de Protection des Personnes, CPP 06/84) approved the study, and all study participants were given complete information and all signed written consent form [17].
Microbiological Analysis of Sputa
Sputum samples were collected by expectoration into a sterile cup after rinsing the mouth with water to prevent excessive salivary contamination and then processed as described in previous studies [17]. Briefly, after pre-treatment, digested sputa were inoculated simultaneously onto ten growth media including: (1) chromogenic medium; (2) in-house Sabouraud glucose peptone agar with 0.5 g/L chloramphenicol and 0.5 g/L cycloheximide; (3) in-house Sabouraud glucose peptone agar with 0.5 g/L chloramphenicol and 0.5 g/L gentamicin; (4) dichloran-rose Bengal chloramphenicol agar supplemented with benomyl; (5) two erythritol-enriched media; (6) chocolate PolyViteX agar; (7) Columbia colistin-nalidixic acid agar; (8) Bromo Cresol Purple agar; (9) blood agar; and (10) Cepacia agar. All media were purchased from bioMérieux (Craponne, France), except Cepacia agar from AES Laboratory (Combourg, France). They were aerobically incubated at 37 °C, except Cepacia agar which was incubated at 30 °C, and the in-house Sabouraud glucose peptone agar and one erythritol agar which were incubated at 27 °C. Colonies growing on these media were identified using conventional methods.
DNA Extraction
Total DNA was extracted from sputa using the High Pure PCR Template Preparation kit (Roche Diagnostics, Meylan, France) as described in other studies [17]. The DNA was then quantified by using PicoGreen DS DNA quantification kit (Molecular Probes, Leiden, The Netherlands).
Molecular Characterization of Bacteria by T-RFLP
Amplification of a conserved region of the 16S rRNA gene of bacteria was performed using the 8f700 oligonucleotide primer labelled at the 5′ end with IRD700, and the reverse unlabelled primer 926r primer, as described in the literature [12]. After PCR amplification, 20 ng of PCR products were digested with the restriction endonuclease CfoI enzyme (Roche, United Kingdom) and then analysed by using a 25-cm SequagelXR denaturing acrylamide gel (National Diagnostics, Hessle, UK) for fragment length separation on a LICOR apparatus (Biosciences, Lincoln, US) as described by Daniel et al. [21]. T-RF band lengths and volumes were determined by comparison with marker microSTEP 15a (700 nm) (Microzone, Lewes, UK) using Phoretix one-dimensional advances software (version 5.10; Nonlinear Dynamics, Newcastle upon Tyne, UK), and expressed as a percentage of the total band lane volume. The resolution region of the method ranged from 50 to 950 bases with a detection threshold of 0.01% of total profile signal, allowing determining the number of T-RF bands detected in each sample and their relative abundance in the four samples.
Molecular Characterization of Fungi by ITS Analysis (ITS–PCR Cloning)
The primers ITS1 and ITS4 [22, 23] were used to amplify the entire internal transcribed spacer rDNA region. A 5 µl-aliquot of each PCR product was cloned using the TOPO-TA cloning kit (Invitrogen, Illkirch, France) followed by transformation into the competent E. coli DH5 alpha cells (pGEM®t easy vector system, JM 109 competent cells, Promega, Charbonnières-les-Bains, France) according to the manufacturer’s recommendations. The presence of the insert in the colonies was checked by real-time PCR amplification using M13 forward (5′-GTAAAACGACGGCCAG-3′) and reverse (5′- CAGGAAACAGCTATGAC -3′) primers. The qPCR reactions were carried out on a LightCycler® 480 (Roche, Mannheim, Germany) in a 20 μl final reaction volume containing 1X LightCycler® 480 SYBR Green I Master (Roche Diagnostics, Meylan, France), 500 nM of upper and lower primers (ITS1–ITS4), and 5 μl of boiled E. coli supernatant. After initial denaturation at 95 °C for 8 min, PCR was performed using 50 cycles at 95 °C for 10 s, 60 °C for 10 s, and 72 °C for 15 s. PCR products were identified by melting curve analysis. Clones showing the same melting curve patterns were grouped in a single taxon, and five representatives of each taxon were sequenced using BigDye Terminator cycle sequencing kit v3.1 protocol (Applied Biosystems, Courtaboeuf, France). Sequences sharing over 99% identity were grouped in the same species or genus.
Fungal and Bacterial UDS Analyses
Two sets of primers were used to amplify the 16S rDNA (V3 region) and ITS2 loci from bacteria and fungi, respectively, as described in the literature [17]. Amplicon libraries were prepared using GS Junior Titanium Kits and pyrosequenced on a GS Junior (Roche/454 Life Sciences, Branford, Connecticut, USA) according to manufacturer’s recommendations (GenoScreen, Lille, France). Data generated from sequencing were trimmed and analysed using the Metabiote® pipeline (GenoScreen). Potential chimeras, reads with poor-quality scores (<20), short reads (<150 for 16S rDNA, <100 for ITS2), homopolymers, singleton or doubleton 16S rDNA reads were removed. OTUs were created applying complete-linkage clustering with 97% similarity criteria (UClust v1.2.22q [24]. The OTUs’ annotation was performed using RDP classifier against Greengenes (release 13_8) and UNITE databases for 16S rDNA and ITS2, respectively, and applying criteria of ≥98% homology with reference sequences and <10−5 e-values [25].
We computed rarefaction curves representing the number of genera (or sections) identified per reads to assess the number of bacterial and fungal taxa expected to be found in the analysed samples, and to evaluate deep sequencing effort in terms of taxonomic diversity findings. We also computed Shannon diversity index and Shannon evenness index to compare the homogeneity in the microbiota composition of the samples. Shannon evenness indicates the distribution of individuals over genera, whereas Shannon diversity index takes into account two features, richness and evenness. The value of the diversity index increases both when richness increases and when evenness increases. For a given number of genera, the value of a diversity index is maximized when all genera are equally abundant.
Statistical Analyses
The comparison of the mean number of genera identified with each method (culture, molecular characterization by PCR-T-RFLP for bacteria and ITS-PCR-cloning for fungi, and UDS analysis) was performed using the Friedman variance analysis test for paired data with a significance level of <0.05.
Results
Samples and Patients
We collected four sputum samples from four CF adult females, aged between 22 and 39, who were part of a long-term follow-up program at “Centre Hospitalier Régional Universitaire de Lille” (MucoFong project) and of whom two were homozygous for the F508 del mutation and two were heterozygous. All were colonized by at least one fungus (yeast or filamentous) during the year before sampling. Patients 2 (P2) and 3 (P3) were treated with inhaled corticosteroids; P2 received long-term azithromycin; P2, P3, and P4 received more than 3 systemic antibiotic courses over the year prior to sampling, and P3 was on per os itraconazole treatment for an allergic bronchopulmonary aspergillosis (ABPA) caused by A. fumigatus (detected in sputum) over the last year. The spirometric parameters (Forced Vital Capacity, FVC; and Forced expiratory volume in one second, FEV1) were low only for P1 and P2. For a healthy patient, FEV1 should be greater than or equal to 90% and FEV1/FVC > 90%.
The basic characteristics of the four patients at enrolment are summarized in Table 1.
Diversity of Microbial Communities as Evaluated by Each Method
Culture using cultures, one or two bacterial species were detected per patient (Figs. 1, 2): Proteus mirabilis and P. aeruginosa for P1; Haemophilus influenzae and P. aeruginosa for P2; Stenotrophomonas maltophilia for P3 and S. aureus for P4. Both mucoid and non-mucoid P. aeruginosa were identified. No mycobacteria were recovered. Fungi detected by culture were Candida albicans, A. fumigatus and Scedosporium apiospermum for P1 and P4, and C. albicans and A. fumigatus for P2 and P3 (Fig. 3).

Boxplot comparison of bacterial (a) and fungal (b) taxonomic diversity in the airway microbiota from four CF patients using three methods: culture, first generation sequencing and ultra-deep sequencing (UDS, 454 technology). The top and bottom boundaries of each box indicate the 75th and 25th quartile values, respectively, and the lines within each box represent the median values. Ends of whiskers mark the lowest and highest diversity values in each instance. The overall comparison of the three methods by Friedman variance analysis test for paired data was significant for both bacteria (p = 0.038) and fungi (p = 0.018). The significance of method-to-method comparisons is shown with asterisks on the plot

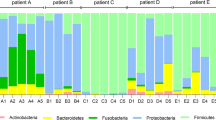
Taxa plots summarising the relative abundance of bacterial genera identified in respiratory samples from four CF patients (P1, P2, P3 and P4) by three methods: culture, first generation sequencing (T-RFLP) and ultra-deep sequencing (UDS, 454 technology)

Taxa plots summarising the relative abundance of fungal genera or sections identified in respiratory samples from four CF patients (P1, P2, P3, and P4) by three methods: culture, first generation sequencing (ITS–PCR cloning—sequencing) and ultra-deep sequencing (UDS, 454 technology)
FGS methods: T RFLP and ITS PCR-cloning for bacteria and fungi detection, respectively, with T-RFLP the bacterial richness was highly variable between patients and ranged from 1 to 8 genera per patient, with a median of four genera (Fig. 1). P1 appeared to be colonized exclusively by Pseudomonas sp. (100%, although Proteus sp. was highly represented in culture, Fig. 2); P2 was colonized by Haemophilus sp. and Pseudomonas sp.; P3 by Pseudomonas sp., Streptococcus sp., Escherichia sp., and several facultative or obligate anaerobes such as Prevotella sp., Veillonella sp., and Rothia sp. However, P4 had the most diversified flora with several anaerobes, Streptococcus sp., Haemophilus sp., Escherichia sp., and Staphylococcus sp. In three out of the four patients (P2, P3, and P4), unidentified bacteria were found by T-RFLP, ranging from 1.5 to 15% of the total taxa. The mean number of genera per patient as detected by T-RFLP was not statistically different from that found by culture (p = 0.56) (Fig. 1).
Using ITS-PCR-cloning, a total of 400 clones containing the correct insert size (approximately 650 bp for the whole ITS region) were collected per patient. For each patient, 100 clones were sequenced and the remaining 300 clones were analysed using melting curves. All sequences exhibited a similarity score of ≥99% when compared with the reference sequences present in the GenBank database. Overall, 1 or 2 fungal genera were identified per patient, which was not statistically different from that revealed by culture (p = 0.16) (Figs. 1, 3).
UDS method using UDS approach, we obtained a total of 15,837 trimmed reads for the bacterial 16S rRNA gene and 18,477 reads for the fungal ITS2 locus. For all samples, the rarefaction curves representing the number of genera (or sections) identified per read reached a plateau, indicating that almost all the microbiota present in each sample were detected. Overall, 99.2% of bacterial reads (clustered in 248 OTUs) and 98.3% of fungal reads (434 OTUs) were attributed to a genus or a section level.
The median number of detected bacterial genera was 11, ranging from 3 to 22 per patient, which was significantly different from findings from conventional approaches (p = 0.038) (Figs. 1, 2). We could not find bacterial genera common to the four patients, and we could not identify a bacterial core (1%) [26], i.e. some bacterial genera were found with a relative abundance of more than 1% in three of the patients. The four patients had highly variable bacterial profiles: P1, P2, and P3 harboured mostly cultivable genera, whereas P4 had over 50% of poorly or non-cultivable genera, since P4 was essentially colonized by anaerobes including Prevotella sp. (23%), Leptotrichia sp. (22.5%) and other anaerobes. This last patient also received 7 antibiotics courses in the previous year, probably eliminating most of the conventional pathogens.
The median number of fungal genera or sections recognised via UDS was 8.5, ranging from 9 to 14, which was statistically different from findings from conventional approaches (p = 0.018) (Figs. 1 and 3). The fungal core was represented by Aspergillus section Fumigati (relative abundance >70% in 3/4 patients) and Penicillium sp. (relative abundance varying from 1.5 to 9% in 4/4 patients). Of note, for P1, A. fumigatus was found by culture but not detected by ITS–PCR cloning or UDS, which could be explained by a possible contamination event during culture (Fig. 3). Candida spp. were detected in all patients (predominantly C. albicans); however, their relative abundance found with UDS for P2 and P3 was low (0.81 and 0.2%, respectively), especially when compared with their relative abundance in culture (6.7 and 28.60%, respectively). Malassezia spp. were found at low relative abundance (0.5%) for P1 and high relative abundance for P3 (17.8%), with a predominance of Malassezia restricta (data not shown).
Analysis of Microbiota Richness According to Lung Function
P1 and P2, who had a severe respiratory disease as assessed by standard spirometry (FEV1 < 50%) (Table 1), exhibited a reduced fungal and bacterial diversity as represented by Shannon equitability and Shannon diversity indexes compared with P3 and P4 (Fig. 4).

Relation between bacterial (a) or fungal (b) richness and lung function expressed as FEV1 and FVC values. Total richness of bacterial or fungal communities from each patient was expressed using Shannon diversity index and is represented by proportional spots of different colours. Red spots represent patients with a highly impaired lung function (P1 and P2), green spots represent a patient with mildly impaired lung function (P3), and blue spots represent a patient with preserved lung function
Comparison of the Three Methods
Overall, 27 bacterial and 18 fungal genera were detected from the four patients. Five (18%) and 3 (16%), 9 (33%) and 3 (16%), 26 (96%) and 18 (100%), were detected by culture, FGS methods, and UDS for bacteria and fungi, respectively. The median number of genera detected per patient by UDS was significantly higher compared with that detected by culture or FGS methods (Fig. 1). On the other hand, no significant difference was noticed between the median number of detected genera by culture and FGS methods. Nonetheless, T-RFLP detected for P3 and P4, who had the highest bacterial diversity, respectively, 4 and 5 additional bacterial genera compared with culture. With culture-based methods, only major cultivable species (e.g. Pseudomonas, Stenotrophomonas, Haemophilus, Staphylococcus, and Proteus for bacteria, and Aspergillus, Scedosporium, and Candida for fungi) were detected. These major species were also detected by the two other methods; however, FGS methods showed limitations and missed Candida spp. in P2, P3, and P4, Scedosporium species in P4, and Proteus species in P1.
Less abundant species and the species that require specific media or conditions to grow were not detected by culture-based methods. For example, the Malassezia, which were detected by UDS, were not detected by culture (Fig. 3). Likewise, anaerobes were detected by T-RFLP and UDS but not by culture (Fig. 2).
Discussion
Although culture-based methods have been widely used for the analysis of bacterial and fungal diversity in respiratory samples of CF patients, these techniques have significant limitations. Few culture-independent strategies have already been developed to replace or complement them [12, 13, 15, 16, 26]. In the present study, we did a comparative analysis of the bacterial and fungal communities compositions and structures of the airway microbiota of four adult CF patients using two culture-independent methods (T-RFLP and ITS-PCR cloning as FGS approach and UDS method), and a conventional culture-based strategy.
Our findings are complementary to those of previous studies [17,18,19, 27, 28], showing major differences between the fungal and bacterial community composition and structure in sputa from CF patients. First, the number of fungal genera per patient was smaller than the number of bacterial genera, which is consistent with the fact that the fungal microbiome (or mycobiome) belongs to the rare biosphere [29]. Every individual’s microbiome contains thousands of microbes, of which 99.9% belong to only a few species, and the less abundant (<0.1%), but more diverse, components of the microbiome constitute the rare biosphere according to Huffnagle and Noverr [29]. Interestingly, this rare biosphere contains less fungal species compared to bacterial species. Secondly, at the individual level, the bacterial microbiota did not demonstrate a high degree of diversity (3–22 genera/patient) and was dominated by a small number of genera with high relative abundance. No bacterial core was identified and each patient had a unique bacterial profile. This was consistent with the results of previous studies showing that the airways of CF patients harbour a reduced bacterial diversity compared to healthy subjects and are associated with a high degree of inter-individual variability and only a small number of common taxa [17, 26, 28, 29]. In contrast, we identified a fungal core since Aspergillus Section Fumigati and Penicillium were present in 75% of the patients with moderate to high relative abundance [17, 30]. This result can easily be linked to the past A. fumigatus colonisation in 3/4 patients during the year prior to sampling.
Depending on the employed method, the assessed bacterial and fungal diversities were significantly different. If all three methods were able to detect the most predominant, easily cultivable genera, as true or potential pathogens, we observed that FGS methods were sometimes unreliable, especially regarding fungi. However, in patients with high bacterial diversity profiles, T-RFLP did better than culture and detected a higher number of genera [12, 13]. Compared with both culture and FGS methods, UDS has proven to be more efficient and more reliable, providing precise taxon identification, even with low abundance (additional 17 bacterial and 15 fungal genera detected compared with FGS methods). Moreover, the bacterial flora was exhaustively identified using UDS with only <0.5% of unidentified sequences versus 5% with FGS methods. As culture relies on a priori selection of organisms with specific culture media and conditions, UDS evidently reveals a much higher diversity for both bacteria and fungi [17]. Apart from the inherent limitations of T-RFLP and ITS-PCR cloning, e.g. T-RFLP is based on amplicon size determination and its resolution power is sometimes too weak to separate organisms that produce very close or similar fragment sizes [13, 31], UDS allows a much higher sequencing depth and therefore detection of less abundant or rare microorganisms. The superior efficiency of UDS versus culture method is easily attributed to the UDS ability to detect uncultivable, difficult to cultivate, or rare microorganisms, like some fungal species [17]. Compared with FGS, UDS efficiency is mostly related to the depth of the new generation sequencing as well as to the inherent limitations of T-RFLP and ITS-PCR cloning.
Among the bacterial genera identified only through molecular methods (22/27), we found, as expected, a variety of obligate or facultative anaerobic genera which are difficult or impossible to detect using standard culture methods. Although the majority of these genera (13/22) were associated with very low relative abundance (<1% per patient), a few of them were markedly represented. For instance, Streptococcus, Prevotella, Veillonella, Leptotrichia, Selenomonas, Rothia, Actinomyces and Porphyromonas were major taxa in the sputa of 2/4 patients. These genera are known as major members of the oral bacterial microbiota [32] and they have already been described as part of a complex signature characterizing the microbial profile of CF airway microbiota [28, 29]. Currently, the role of anaerobic bacteria is not well understood in CF lower airway disease. Studies have shown that the relative abundance of anaerobes fluctuates in the lower airways of CF patients, and that a reduced lung function and increased inflammatory reaction were associated with a decrease in anaerobic load [8, 33].
Among the fungal taxa identified via molecular methods only, eight were not rare taxa (relative abundance >1% in at least one patient). Two of these genera (Penicillium and Cladosporium) are environmental fungi, often isolated from respiratory secretions by cultures; two (Malassezia and Saccharomyces) are transient or persistent human commensal yeasts; one (Bjerkandera) is both a wood-rotting macromycete and an agent of asthma exacerbation; and the three others (Gloeotinia, Parastagonospora, Pseudopithomyces) are cereals pathogenic fungi. Except for the cereals fungi, which probably have a food-borne origin, the other fungi are known to be part of the CF airway microbiota [17, 19, 32, 33]. Unlike bacteria, these fungal genera (except Malassezia) are cultivable using standard Sabouraud dextrose agar. However, in CF patients, the conventional fungal cultures are sometimes subjected to bacterial overgrowth mainly by predominant bacteria, such as P. aeruginosa, which can hinder the growth of some fungi, thus explaining the lower efficiency of culture method to isolate the whole fungal biota.
Briefly, UDS is a powerful method to identify dominant taxa, estimate their diversity, and detect minor or difficult-to-cultivate taxa, such as Malassezia spp. or anaerobes. As it has been described previously, about 60% of the species inhabiting the airways of CF patients are not detected by cultures; thus, UDS appears to be a suitable tool for further exploration of CF respiratory flora [17]. Furthermore, UDS methods are able to discriminate the fungal taxonomic level genera and sometimes the level of the section, such as Fumigati or Flavi, whereas culture-based methods do not give these precisions that are important in the therapeutic management of patients.
However, UDS methods also have inherent bias sources, mostly related to extraction protocols or choice of amplification targets, which can lead to misrepresentation of some bacterial or fungal taxa [34,35,36]. In our study, for instance, the identification of major cultivable taxa concurred between culture-based method and UDS; however, we observed significant variations in terms of relative abundance between the two methods. In one patient, we found 50% of Proteus via culture, whereas UDS detected this genus at a low abundance (<1%). To fully apprehend the CF airway microbiota, its disturbances, and the clinical manifestations associated with, it is vital to get a true picture of the bacterial and fungal community structures. For this purpose, minimizing UDS biases and thus defining robust standardized protocols, especially regarding extraction protocols and PCR amplification target, will be of major interest. Different extraction protocols, mostly adapted for stool samples and the corresponding gut microbiota analysis, have been tested for bacterial diversity assessment [37], but few studies have focused on protocols dedicated to both bacterial and fungal extractions, especially in respiratory specimens [38]. Available data highlighted the great variation of the relative abundance of the major and minor fungal taxa from one extraction protocol to another, whereas the bacterial community structure appeared more stable (Angebault et al. Poster 0977, ECCMID 2017) [35]. In terms of fungal target choice, ITS1 is the most commonly used [38]. However, its added value compared with ITS2 has not been demonstrated yet. In addition to the technical optimization of UDS method, the metagenomic approach should probably be used in combination with (rather than instead of) an optimized version of culture, such as the culturomics approach. The latter is based on the use of multiple culture media under various conditions (temperature, duration of incubation) to assess, as exhaustively as possible and without the UDS biases, the microbiota [39, 40]. Moreover, maintaining a culture-based approach will also allow assessing antibiotic and antifungal susceptibility testing of patient isolates, an essential step in patient’s management. Finally, UDS allows the simultaneous detection of fungi and bacteria which is rarely done. But the cost, time and analysis capacity needed for UDS are not appropriate for routine use in clinical microbiology laboratories.
In conclusion, our study, despite the limitations related to the small number of enrolled patients, highlighted benefits on UDS for fungal and bacterial population detection. With UDS method, we were able to detect taxa, not revealed by culture or FGS methods, at relatively high proportions, and to designate a mycobiota core composed of Aspergillus belonging to the Fumigati section and Penicillium sp. which was consistent with the patient past history of A. fumigatus colonisation the year before. Similarly to other studies [17, 27], we observed a reduced diversity of fungal and bacterial communities in patients with impaired lung functions (low FEV1 and FCV). These data reinforce the major interest of using well-characterized UDS approach in combination with other appropriate methods in clinical laboratories to fully analyse the CF airway microbiota, to understand the ecological processes of CF airway microbiota during non-exacerbation periods, and to open new path towards better CF healthcare oriented to halt fungi or bacteria-related disease progression. Beside these bacterial—fungal interactions, it is also likely that other agents, such as viruses, play an important role in the CF airway microbiota [41].
References
Vaincre la Mucoviscidose. Registre français de la mucoviscidose - Bilan des données 2015. [Internet]. 2015 [cited 2014 Apr 11]. Available from: http://www.vaincrelamuco.org/sites/default/files/registre_francais_de_la_mucoviscidose_-_bilan_2015_v4.pdf.
Waters V, Ratjen F. Antibiotic treatment for nontuberculous mycobacteria lung infection in people with cystic fibrosis. Cochrane Database Syst Rev. 2016;12:CD010004.
Salvatore D, Buzzetti R, Mastella G. An overview of international literature from cystic fibrosis registries. Part 5: Update 2012-2015 on lung disease. Pediatr Pulmonol. 2016;51:1251–63.
European Cystic Fibrosis Society. ECFS Patient Registry Annual Data Report 2014 [Internet]. 2014 [cited 2017 Apr 11]. Available from: https://www.ecfs.eu/projects/ecfs-patient-registry/annual-reports.
LiPuma JJ. The changing microbial epidemiology in cystic fibrosis. Clin Microbiol Rev. 2010;23:299–323.
Huang YJ, LiPuma JJ. The microbiome in cystic fibrosis. Clin Chest Med. 2016;37:59–67.
Parkins MD, Floto RA. Emerging bacterial pathogens and changing concepts of bacterial pathogenesis in cystic fibrosis. J Cyst Fibros. 2015;14:293–304.
Tunney MM, Field TR, Moriarty TF, et al. Detection of anaerobic bacteria in high numbers in sputum from patients with cystic fibrosis. Am J Respir Crit Care Med. 2008;177:995–1001.
Borman AM, Palmer MD, Delhaes L, et al. Lack of standardization in the procedures for mycological examination of sputum samples from CF patients: a possible cause for variations in the prevalence of filamentous fungi. Med Mycol. 2010;48:S88–97.
Williams C, Ranjendran R, Ramage G. Pathogenesis of fungal infections in cystic fibrosis. Curr Fungal Infect Rep. 2016;10:163–9.
Sudfeld CR, Dasenbrook EC, Merz WG, Carroll KC, Boyle MP. Prevalence and risk factors for recovery of filamentous fungi in individuals with cystic fibrosis. J Cyst Fibros. 2010;9:110–6.
Rogers GB, Carroll MP, Serisier DJ, et al. Characterization of bacterial community diversity in cystic fibrosis lung infections by use of 16S ribosomal DNA terminal restriction fragment length polymorphism profiling. J Clin Microbiol. 2004;42:5176–83.
Rogers GB, Skelton S, Serisier DJ, van der Gast CJ, Bruce KD. Determining cystic fibrosis-affected lung microbiology: comparison of spontaneous and serially induced sputum samples by use of terminal restriction fragment length polymorphism profiling. J Clin Microbiol. 2010;48:78–86.
Suhr MJ, Banjara N, Hallen-Adams HE. Sequence-based methods for detecting and evaluating the human gut mycobiome. Lett Appl Microbiol. 2016;62:209–15.
Armougom F, Bittar F, Stremler N, et al. Microbial diversity in the sputum of a cystic fibrosis patient studied with 16S rDNA pyrosequencing. Eur J Clin Microbiol Infect Dis. 2009;28:1151–4.
Guss AM, Roeselers G, Newton ILG, et al. Phylogenetic and metabolic diversity of bacteria associated with cystic fibrosis. ISME J. 2011;5:20–9.
Delhaes L, Monchy S, Fréalle E, et al. The airway microbiota in cystic fibrosis: a complex fungal and bacterial community–implications for therapeutic management. PLoS ONE. 2012;7(4):e36313.
Kramer R, Sauer-Heilborn A, Welte T, et al. Cohort study of airway mycobiome in adult cystic fibrosis patients: differences in community structure between fungi and bacteria reveal predominance of transient fungal elements. J Clin Microbiol. 2015;53:2900–7.
Willger SD, Grim SL, Dolben EL, et al. Characterization and quantification of the fungal microbiome in serial samples from individuals with cystic fibrosis. Microbiome. 2014;2:40.
Mahboubi MA, Carmody LA, Foster BK, et al. Culture-based and culture-independent bacteriologic analysis of cystic fibrosis respiratory specimens. J Clin Microbiol. 2016;54:613–9.
Daniels TWV, Rogers GB, Stressmann FA, et al. Impact of antibiotic treatment for pulmonary exacerbations on bacterial diversity in cystic fibrosis. J Cyst Fibros. 2013;12:22–8.
White TJ, Bruns L, Lee S, Taylor J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protocols: A Guide to Methods and Applications. New York: Academic Press; 1990. p. 315– 322.
Sitterlé E, Rodriguez C, Mounier R, et al. Contribution of ultra deep sequencing in the clinical diagnosis of a new fungal pathogen species: Basidiobolus meristosporus. Front Microbiol. 2017;8:334.
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–1.
Wang Q, Garrity GM, Tiedje JM, Cole JR. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–7.
van der Gast CJ, Walker AW, Stressmann FA, et al. Partitioning core and satellite taxa from within cystic fibrosis lung bacterial communities. ISME J. 2011;5:780–90.
Coburn B, Wang PW, Diaz Caballero J, et al. Lung microbiota across age and disease stage in cystic fibrosis. Sci Rep. 2015;5.
Blainey PC, Milla CE, Cornfield DN, Quake SR. Quantitative analysis of the human airway microbial ecology reveals a pervasive signature for cystic fibrosis. Sci Transl Med. 2012;4:153ra130.
Huffnagle GB, Noverr MC. The emerging world of the fungal microbiome. Trends Microbiol. 2013;21:334–41.
Nguyen LDN, Viscogliosi E, Delhaes L. The lung mycobiome: an emerging field of the human respiratory microbiome. Front Microbiol. 2015;6:89.
Zakharkina T, Heinzel E, Koczulla RA, et al. Analysis of the airway microbiota of healthy individuals and patients with chronic obstructive pulmonary disease by T-RFLP and clone sequencing. PLoS ONE. 2013;8(7):e68302.
Aas JA, Paster BJ, Stokes LN, Olsen I, Dewhirst FE. Defining the normal bacterial flora of the oral cavity. J Clin Microbiol. 2005;43:5721–32.
Sherrard LJ, Bell SC, Tunney MM. The role of anaerobic bacteria in the cystic fibrosis airway. Curr Opin Pulm Med. 2016;22:637–43.
Kashinskaya EN, Andree KB, Simonov EP, Solovyev MM. DNA extraction protocols may influence biodiversity detected in the intestinal microbiome: a case study from wild Prussian carp, Carassius gibelio. FEMS Microbiol Ecol. 2017;93(2). pii:fiw240.
Vesty A, Biswas K, Taylor MW, Gear K, Douglas RG. Evaluating the impact of DNA extraction method on the representation of human oral bacterial and fungal communities. PLoS ONE. 2017;12(1):e0169877.
Rogers GB, Bruce KD. Next-generation sequencing in the analysis of human microbiota: essential considerations for clinical application. Mol Diagn Ther. 2010;14:343–50.
Doré J, Ehrlich SD, Levenez F, et al. IHMS_SOP 06 V1: Standard operating procedure for fecal samples DNA extraction, Protocol Q. [Internet]. International Human Microbiome Standards; 2015. Available from: http://www.microbiome-standards.org.
Schoch CL, Seifert KA, Huhndorf S, et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci USA. 2012;109:6241c6.
Lagier J-C, Armougom F, Million M, et al. Microbial culturomics: paradigm shift in the human gut microbiome study. Clin Microbiol Infect. 2012;18:1185–93.
Abdallah RA, Beye M, Diop A, et al. The impact of culturomics on taxonomy in clinical microbiology. Antonie Van Leeuwenhoek. 2017;. doi:10.1007/s10482-017-0871-1.
Vallet S. Viruses in cystic fibrosis patients’ airways. Crit Rev Microbiol. 2017;24:1–19.
Stevens DA, Moss RB, Kurup VP, et al. Allergic bronchopulmonary aspergillosis in cystic fibrosis–state of the art: cystic Fibrosis Foundation Consensus Conference. Clin Infect Dis. 2003;37(Suppl 3):S225–64.
Acknowledgements
We would like to thank Damian Rivett and Jean-Philippe Barnier for their technical assistance, and Genoscreen Society for their critical and technical support.
Funding
This work was supported in part by the Assistance Publique-Hôpitaux de Paris (public sector, bourse de mobilité), and Pfizer France Pharmaceutical Division. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. L. Delhaes has received research grants from the French Ministry of Health and Research (PHRC N°2006/1902), Centre Hospitalier Régional Universitaire de Lille, the association “Vaincre la Mucoviscidose” (MucoFong and Mucofong-ATF N8 2006/351), and Pfizer France Pharmaceutical Division (Nu 2006/158).
Author information
Authors and Affiliations
Contributions
Authors’ Contributions
FW, BW and LD collected sputum samples and clinical data. FB, OC, JMC, FS and LD prepared sputum samples for molecular analysis including UDS. CA performed the statistical analyses including that of UDS. FB and LD contributed to study design. FB was in charge of all the molecular approaches. FB and CA wrote the manuscript with significant contributions of co-authors, especially KB and LD. All authors read and gave approval on the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
FB received grants from Astellas, Pfizer, and payment for lectures from Merck and Gilead.
Ethical Approval
All procedures performed in this study involving human participants were in compliance with the ethical standards of the institutional research committee (Comité de Protection des Personnes of Lille University Hospital, CPP 06/84) and with the 1964 Helsinki Declaration and its latest amendments.
Informed Consent
Informed consent forms were obtained from all individual participants included in the study.
Rights and permissions
About this article

Cite this article
Botterel, F., Angebault, C., Cabaret, O. et al. Fungal and Bacterial Diversity of Airway Microbiota in Adults with Cystic Fibrosis: Concordance Between Conventional Methods and Ultra-Deep Sequencing, and Their Practical use in the Clinical Laboratory. Mycopathologia 183, 171–183 (2018). https://doi.org/10.1007/s11046-017-0185-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11046-017-0185-x