
Abstract
Background
Upland cotton is one of the utmost significant strategic fiber crops, and play a vital role in the global textile industry.
Methods and results
A total of 128 genotypes comprised Gossypium hirsutum L, Gossypium barbadense L., and pure lines were used to examine genetic diversity using iPBS-retrotransposon markers system. Eleven highly polymorphic primers yielded 287 bands and 99.65% polymorphism was recorded. The mean polymorphism information content was estimated at 0.297 and the average diversity indices for the effective number of alleles, Shannon’s information index, and overall gene diversity were 1.481, 0.443, and 0.265, respectively. The analysis of molecular variance (AMOVA) revealed that 69% of the genetic variation was within the population. A model-based STRUCTURE algorithm divided the entire germplasm into four populations and one un-classified population, the genotypes G42 (originating in Egypt) and G128 (originating in the United States), showed the highest genetic distance (0.996) so these genotypes could be suggested for breeding programs as parental lines.
Conclusions
This is the first investigation using an iPBS-retrotransposon marker system to examine the genetic diversity and population structure of upland cotton germplasm. The rich diversity found in upland cotton germplasm could be exploited as a genetic resource when developing breeding programs and could also help with efforts to breed cotton around the world. These findings also show the applicability and effectiveness of iPBS-retrotransposons for the molecular characterization of cotton germplasm.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cotton fiber is a well-known and leading textile fiber in the textile industry, with an annual economic impact of $600 billion worldwide [1]. The fiber is produced from cotton plants. These plants are perennial woody shrubs and trees in their wild form, but cultivated cotton is primarily an annual plant [2]. Cotton belongs to the family Malvaceae and the genus Gossypium, a genus of nearly 50 species worldwide, from tropical, subtropical, and semi-arid regions. There are currently four cultivated species of cotton of which two are diploids (2n = 2x = 26) and two are allotetraploids (2n = 4x = 52). Eight genome types (from A to G and K) are found in cotton (Gossypium spp.). Diploid (AA) cultivated species are G. herbaceum and G. arboretum, while allotetraploid (AADD) cultivated species are G. hirsutum and G. barbadense [3,4,5]. The most globally produced species of cotton are allotetraploid, particularly G. hirsutum also known as upland cotton. G. hirsutum accounts for almost 96–97% while G. barbadense accounts for 2–3% of the total worldwide cotton cultivation area. Only 1% of the world’s cotton farmland is used to grow G. arboreum and G. herbaceum [3, 4, 6]. Because of the increased need for food, feed, fiber, and fuel as a result of the increasing world population which is estimated to reach 11 billion in 2050. It is critical to improving agricultural production by at least two to three times. Multiple biotic and abiotic stresses produced due to climate change also caused a decline in global agricultural production. These concerns highlight the urgent need to increase crop productivity. In addition to addressing such limited resources, it is critical to make effective use of existing genetic diversity [7].
Due to the economic importance of cotton in several countries, cotton breeding programs are primarily focused on improving fiber quality yield, as well as early maturity and pest and disease resistance. Breeders are working to increase drought, heat, and salt stress tolerance in cotton varieties to help mitigate the effects of climate change and global warming [6, 8]. The world cotton fiber production rate in the 2019–2020 season was increased slightly (approximately 1%) compared to the previous year and reached 25,929 thousand tons, and it has been confirmed that it is 24,800 thousand tons in 2020–2021 (Dec.). India, which surpasses China in cotton production in recent years, took first place in cotton production by providing approximately 23% of the world’s production in the 2019–2020 season. India is followed by China, the USA, and Brazil [9, 10]. Meanwhile, the cotton fiber production in Turkey in the 2019–2020 season fell to 751 thousand tons with a descending compared to the previous season and after Uzbekistan took place in seventh place[10]. Turkey is present in the list of top 10 cotton-producing countries, which ranks eighth after India, China, the United States of America, Pakistan, Brazil, Australia, and Uzbekistan but still, its consumption demand is higher than domestic production. As a result, Turkey imports 50% of its cotton which makes it one of the major cotton importers after Bangladesh, Vietnam, and China [1, 6]. Cotton is a summer cash crop (sown in March/April and harvested in September/October), that is cultivated in three main regions of Turkey: the Aegean, Mediterranean (mainly Antalya and Cukurova), and Southeastern Anatolia [6, 11]. Cotton faces several challenges and constraints in these regions, resulting in low lint yield and fiber quality. One of them is the extreme heat stress (temperatures above 30 °C) in the Southeastern Anatolia region around mid-July and mid-August. Furthermore, in major irrigated agricultural areas, poor irrigation management has resulted in significant salt accumulation in soils [6]. During the hot and dry summer in the Aegean region, irrigation is a major challenge in cotton cultivation, especially for G. hirsutum L [12]. The above-mentioned circumstances and challenges are urging breeders to develop drought, heat, and salt-tolerant cotton varieties.
The abundance of genetic diversity in germplasm resources is critical to the success of any breeding program. The knowledge of genetic relationships among plant genotypes is essential for getting useful insights into the complexity of existing germplasm, uncovering genotype differences, and developing sustainable germplasm conservation plans. It is particularly difficult to create molecular markers for cotton due to its lower level of intraspecific DNA polymorphism. Thus, cotton’s tetraploid genome is relatively huge, containing 2200–3000 Mb of DNA [8]. Molecular markers that have been used to evaluate cotton diversity include AFLP, ISSR, RAPD, and SSR [3, 13,14,15,16,17]. The latest advances in molecular markers and genome sequencing have created a huge potential for assessing genetic diversity in a vast germplasm collection. For instance, inter-primer binding site (iPBS) markers have recently become the most important and widely used marker system for determining genetic diversity and relationships in numerous plants [18]. It has been successfully used in a wide range of genetic diversity studies [19,20,21,22,23,24,25,26,27]. This PCR-based marker system was developed by [18] based on transposon elements, particularly the long terminal repeat retro-transposons (LTR). One of its benefits is that it does not need knowledge of the genome sequence. Practically, the tRNA complement by its 3’ terminal sequences binds to fully conserved primer binding sites (PBSs) of retrotransposons close to the 5’ LTR. Consequently, the reverse transcription process begins for sequence amplification. Because of its effectiveness in PCR amplification and capacity to screen broad sections of plant genomes, this dominant marker system is ideal for use in genetic diversity analyses for a large number of plant genotypes [21, 29]. This research aimed to explore the genetic diversity of cotton germplasm using the iPBS-retrotransposon marker system, which will contribute to determining population structure and easing the task of cotton breeders, particularly Turkish breeders, in the context of abiotic stresses.
Materials and methods
Plant material
The cotton plant materials used in this study were from the genetic collections of the Nazilli Cotton Research Institute, which included 110 cotton genotypes, and the genetic collections of the University of Dicle - Faculty of Agriculture (Diyarbakir, Turkey), which had 18 cotton pure lines. There were a total of 128 genotypes from various countries (Fig. 1), the majority of which originated in Turkey and the United States. Brief details about the plant material is given in Table 1. The entire cotton germplasm was sown in pots under greenhouse conditions, and young, fresh and undamaged leaves were picked for DNA extraction.

Map showing the collection countries of investigated upland cotton germplasm
DNA isolation
Young, healthy and fresh leaves were collected at a precise time for the extraction of genomic DNA. CTAB protocol proposed by [30] was used for DNA extraction, with minor modifications by [31]. The concentration of DNA was evaluated with the help of a MaestroNano Pro spectrophotometer (MN-913 A, MaestroGen Inc., Hsinchu City, Taiwan – R.O.C.) and further confirmed by agarose gel electrophoresis (i.e. 0.8% agarose gel). The final DNA concentration for the 128 accession samples to be used in polymerase chain reactions (PCR) was adjusted to 5 ng/µL; the samples were stored at -25 oC till the start of PCR amplifications.
iPBS-retrotransposon assessment
In total, 63 different iPBS-retrotransposon primers,taken from the work of [28], were screened for polymorphism using four randomly picked cotton genotypes. Consequently, for the genetic diversity assessment of all 128 cotton genotypes, only eleven of the most polymorphic primers with perfect, clear and visible banding patterns were chosen. The selected iPBS-retrotransposon primers, as well as their annealing temperatures and sequences, are listed in Table 2. For PCR amplification, Kalendar et al. [28]’ protocol was employed. A volume of 20 µL of PCR reaction mixture was prepared, to which 5 ng of template DNA, as well as a PCR mix, were added. The PCR mix consisted of 1X PCR buffer (Thermo Scientific), 0.6 mM for 18-nt primers or 1 mM primer for 12–13-nt primers, 0.2 mM dNTP for each type of deoxyribonucleotide triphosphate (Thermo Scientific, Waltham, MA, USA), 2 mM MgCl2, and 0.2 U Taq DNA polymerase (Thermo Scientific). For PCR, a pre-denaturation stage at 95 °C for 3 min was performed, followed by 30 cycles, which included a denaturation stage at 95 °C for 15 s, an annealing stage with a temperature of 50–65 °C (primer-dependent) for 1 min, and a terminal extension stage at 72 °C for 5 min. The agarose gel electrophoresis (2%, w/v) and containing 0.5 × Tris-borate-EDTA (TBE) buffer was performed to run PCR amplicons for 155 min. Then, ethidium bromide was carefully utilized to stain the gel for a finer visualization with the Imager Gel Doc XR + system (Bio-Rad, Hercules, CA, USA). A ladder (100 bp+) was used as a molecular weight marker.
Data analysis
Since only strong, clear and distinct bands were considered for scoring. Based on the dominant character of the iPBS-retrotransposon marker system, a binary scoring system was adopted in this study. As a result, PCR products were given a score of 0 for the absence and a 1 for the presence of clear and net bands. A 100 bp DNA ladder was used to estimate band sizes. POPGENE software (VER 1.32) was used to compute diversity indices including Shannon’s information index (I), the effective number of alleles (Ne), gene diversity (He), and Nei’s genetic distance [32]. The polymorphism information content (PIC) was estimated by applying the formula by [33] “PIC = 2 fi (1-fi)”), with fi representing the frequency of present loci and (1-fi) for the absent loci of a molecular marker. The principal coordinate analysis (PCoA) and analysis of molecular variance (AMOVA) were executed using GeneALEx version 6.5 software [34]. R statistical software (ver.3.4.1) was used to construct an unweighted pair-group arithmetic mean (UPGMA). The Bayesian clustering approach was utilized to learn more about the genetic structure of the entire cotton germplasm by using the STRUCTURE software (VER 2.3.4, Stanford, CA, USA). The most favorable number of clusters (K subpopulations) was calculated using a protocol proposed by [35], in which 10 independent runs were adjusted for each K value. Thus, also for each run, the preliminary burn-in phase was adjusted to 50,000 with 100,000 MCMC (Markov chain Monte Carlo) iterations. Afterward, the logarithm probability relative to the standard deviation (ΔK) was plotted against the number of clusters (K). The last categorization of specific genotypes was about the proportion of the membership coefficient, which was larger than or equal to 75% (Q ≥ 75%).
Results of the genetic diversity and population distribution assessment
All eleven polymorphic iPBS-retrotransposon primers utilized in this investigation yielded a total of 287 bands with perfect bounding features. The average number of bands per primer was observed 26, furthermore the following primers, 2385 and 2384, displayed the maximum and minimum numbers of bands at 35 and 18, respectively. Out of these 287 total strong and visible bands, 286 (99.65%) were found to be polymorphic with an average yield (26) per primer while the range of polymorphism was 96–100% (Table 3). The mean PIC measured value was 0.297, which ranged from 0.340 (2277) to 0.240 (2386), respectively. The same primers, 2277 and 2386, had the highest (1.579) and lowest (1.318) effective number of alleles (Ne) respectively, with an average of 1.481. Primer 2277 and primer 2386 exhibited the highest (0.502) and lowest (0.336) Shannon’s information index values, respectively. Moreover, the mean for Shannon’s information index was 0.443. The maximum and minimum genetic diversity values of 0.336 and 0.208 were recorded for primers 2277 and 2386, respectively. A similar observation was observed in overall gene diversity as primer 2277 found a maximum (0.315), while 2386 showed a minimum (0.155), respectively with a mean of 0.265 (Table 3).
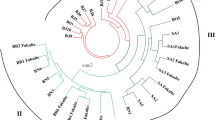
The genetic distance (GD) was found among the whole germplasm (128 accessions) of cotton to understand the clear image of genetic variability. The greatest Nei’s genetic distance (0.996) was found between genotypes 42 (GS-363) and 128 (GS-756), while the lowest genetic distance (0.0574) was found between genotypes 125 (GS-753) and 126 (GS-754). The AMOVA revealed 31% variation among populations and 69% within populations in the safflower germplasm (Table 4).The most favorable number of clusters (K subpopulations) was estimated by observing the position of the sharp peak at k = 4 (Fig. 2). The model-based structure divided the entire cotton germplasm into four distinct populations; 35 genotypes (27.34 of the total) in Pop A, 27 genotypes (21.09%), 29 genotypes (22.66%), 24 genotypes (18.75%) in Pop B, C and D, respectively, based on membership coefficients greater than or equal to 75%. Those genotypes that have a low membership coefficient were categorized into an un-classified population (Fig. 3). The Neighbour -Joining clustering split all 128 accessions into 2 main groups A and B. Group A contained twenty-eight accessions, whereas rest of the all 100 accessions were clustered in group B. Group B further categorized into 2 subgroups, B1 and B2, with ninety three and seven accessions respectively (Fig. 4). The PCoA also separated all accessions into distinguished groups (Fig. 5).

Delta K (ΔK) showing the number of populations resulted through the application of iPBS-retrotransposons

Population structure of upland cotton germplasm as determined by the iPBS-retrotransposons marker system

Neighbor-Joining analysis of upland cotton germplasm revealed by iPBS-retrotransposons markers

Principal coordinate analysis (PCoA) for upland cotton germplasm using iPBS-retrotransposons markers
Discussion
Polymorphism level in cotton germplasm based on iPBS-retrotransposons markers
Several researchers have carried out cotton germplasm characterization in various regions of the globe. To the best of our knowledge, none of them have used retrotransposon markers. iPBS-retrotransposon markers exhibited a high percentage of polymorphic bands in comparison with other molecular markers employed in previous genetic diversity studies on cotton, such as AFLP markers [36], RAPD [37,38,39,40], EST-SSRs [41], SSRs [40], ISSRs [39, 40]. In this study, the average polymorphic band per primer is greater compared to the 2.33 recorded by [41] when they utilized microsatellite markers (SSR) to assess numerous Chinese cotton accessions. It was also higher than the 2.13, 3.6, 3.6 and 4.2 average bands/primer obtained by de [42,43,44,45], respectively, where they used SSR markers to investigate the genetic relationship among various cotton cultivars in their different studies. The obtained average polymorphic bands per primer were also greater than that revealed by [41] using EST-SSR markers to assess the genetic relationship of various cotton cultivars from China (5.08 bands/primer). Moreover, the obtained mean bands/primer were also higher as compared to the 4.7 reported by [37], who used RAPD markers to investigate the genetic diversity of numerous genotypes of elite cotton in Egypt. [17] used ISSR markers to analyze genetic diversity among 22 Turkish cotton (G. hirsutum L.) genotypes and found a lower average number of bands/primers of 17.2.
The polymorphic information content (PIC) results obtained in the present investigation was slightly lower than the 0.40 reported by de [42] when they investigated the genetic relationship among numerous Brazilian cotton (G. hirsutum L.) cultivars using SSRs markers and the 0.46 reported by [46] when they assessed the genetic relationship of several upland cotton (G. hirsutum L.) cultivars using mapped SSRs markers. Similarly, it was lower than the 0.62 by [43] when they assessed genetic diversity among several different upland cotton genotypes from different ecological regions of China using SSR markers and the 0.80 reported by [41] when they investigated genetic diversity among several cotton (G. hirsutum L.) cultivars from China’s main cotton-growing regions using EST-SSR markers. In this current study, iPBS retrotransposon markers displayed a mean PIC value of 0.297. This PIC value was slightly superior compared to the 0.18 uncovered by [15] when they analyzed the genetic relationship among glandless upland cotton germplasm using AFLP markers. Thus, PIC values of 0.25 to 0.5 describe loci of moderate polymorphism [47]. Consequently, these suggest that iPBS retrotransposon markers are effective for cotton genetic relationship studies concerning the resultant intermediate PIC value. It was also reported by [48] that markers with superior PIC values are better at detecting polymorphism in a particular population.
Analysis of genetic diversity and population distribution
The presence of genetic variations among this cotton germplasm was demonstrated by estimating various diversity indices (Table 3). The average effective number of alleles (Ne) found in the present study was slightly higher than the mean Ne value (1.4432) revealed by [17] when they investigated genetic relationship among 22 Turkish cotton (G. hirsutum L.) genotypes using ISSRs markers and slightly higher than 1.413 obtained by [40] also employing ISSR markers to study genetic relationship in the Opal cotton (Gossypium hirsutum L.) germplasm. Furthermore, the obtained Ne value was also higher than the 1.354 reported by [46] when they assessed the genetic relationship of 51 upland cotton cultivars with mapped SSR markers. A Ne value of 1.338 was reported by [39] when they studied genetic diversity among Mehr (Gossypium hirsutum L.) cotton progenies and the parent cultivars using ISSR markers. [40] have reported inferior Ne values by employing SSR and RAPD in a combined genetic diversity study on the Opal cotton (Gossypium hirsutum L.) germplasm with Ne values of 1.249 and 1.208, respectively. A smaller Ne value (1.12) was reported by [49] when they determined genetic diversity in 21 cotton genotypes by using EST-SSR markers. By contrast, the obtained Ne value by employing iPBS-retrotransposons markers was beneath the Ne value (1.7391) reported by Liu et al. [38] when they examined the genetic relationship of 21 accessions of (Gossypium hirsutum L.) by employing RAPD markers. The Ne value was also lower than the 1.514 reported by [39] when they investigated genetic diversity among Mehr (Gossypium hirsutum L.) cotton progenies and the parent cultivars using RAPD markers.
Mean Shannon’s information index was found a higher than the 0.4199 revealed by [17] when they assessed genetic diversity among 22 Turkish cotton (G. hirsutum L.) genotypes using ISSR markers. It was also higher than the 0.321 reported by [46] when they studied the genetic diversity of 51 upland cotton cultivars with mapped SSR markers. Moreover, the obtained I value was higher (0.318, 0.250, and 0.163) as reported by [40] in a combined genetic diversity study on the Opal cotton (G. hirsutum L.) germplasm with the use of ISSR, SSR, and RAPD markers, respectively. Moreover, [39] reported inferior I values in a combined study on genetic diversity among Mehr (G. hirsutum L.) cotton progenies and the parent cultivars using RAPD (0.435) and ISSR (0.290) markers. A smaller mean value of Shannon’s information index (0.10) was reported by [49] when they investigated genetic diversity in 21 cotton genotypes by using EST-SSR markers. However, [38] reported a greater mean value (0.5773) of Shannon’s information index when they analyzed the genetic diversity of 21 accessions of G. hirsutum L. by employing RAPD markers.
In this study, the obtained mean value of gene diversity was higher than the 0.2725 reported in the study by [17] when they evaluated genetic diversity among 22 Turkish cotton (G. hirsutum L.) genotypes using ISSR markers. During the assessment of the genetic diversity of opal cotton (G. hirsutum L.), higher H values i.e., 0.223, 0.154, 0.114 were observed by [40] using ISSR, SSR and RAPD respectively.
A smaller mean H value (0.196) was reported by [39] when they analyzed the genetic relationship among Mehr (Gossypium hirsutum L.) cotton progenies and the parent cultivars using ISSR markers. On the other hand, a higher mean value of genetic diversity (0.4017) was reported by [38] when they analyzed the genetic diversity of 21 accessions of G. hirsutum L. by employing RAPD markers. Additionally, when [39] used RAPD markers to investigate genetic diversity among Mehr cotton (Gossypium hirsutum L.) progenies and parent cultivars, they found a slightly higher mean H value (0.294).
The existence of larger values for different diversity indices (Ne, I, H) with few exceptions could be attributed to the greater effectiveness of the iPBS-retrotransposon marker system to assess the genetic relationships, or it might also be due to the size and nature of the cotton genotype collection. The recorded mean genetic distance among 128 cotton genotypes was 0.325238. Two genotypes, G42 (originated from Egypt) and G128 (originated from the USA), exhibited the highest genetic distance (0.996). They can be suggested as potential parents in prospective cotton breeding studies since they are genetically different. The lowest genetic distance was observed between G125 (originated from the USA) and G126 (originated from Turkey) showed suggesting a genetic relationship. Historical facts might provide a plausible explanation here. In the 1930s, top cotton research and breeding institutions from the United States and Turkey exchanged several cotton genotypes [17]. The analysis of molecular variance (AMOVA) was used to determine the pattern of the partition of the total gene diversity among and within populations and to assess genetic differentiation. AMOVA showed that most of genetic variations within populations.
The clustering techniques STRUCTURE, principal coordinate analysis (PCoA), and neighbor-joining analysis have been used to elucidate the genetic diversity and population structure of various crops. STRUCTURE algorithm has shown more robustness in earlier research studies [50,51,52]. Therefore, in this work, the structure is used as a benchmark for cluster analysis. STRUCTURE algorithm divided the entire germplasm into four distinct populations (Pop A to Pop D) (Fig. 2). Each accession was categorized into a precise cluster population according to its individual ancestry coefficient, which was set at Q ≥ 75%. Thus, Population A is made up of admixtures of genotypes from a variety of countries, primarily Turkey (18 entries), followed by genotypes from the United States, which have eight individuals. Furthermore, Australia has four entries; Pakistan has two, while Greece, Bulgaria, and Egypt each have one entry. Population B consisted of genotypes from seven different countries, mostly Turkey and the United States, which have ten and eight entries, respectively, followed by Australia and Bulgaria, which have four and two entries, respectively. Finally, Syria, Spain, and Azerbaijan each got one entry. Population C mainly consisted of cotton genotypes from Turkey (16 genotypes) and 3 genotypes from the USA. Moreover, all cotton pure lines (18 genotypes) from Turkey also are grouped in this population, whereas in population D, there is an admixture of genotypes originated from 6 different countries. The USA and Turkey are leading with nine and eight genotypes, respectively. Greece and Uzbekistan got 2 genotypes each, and lastly, countries like Iran and Tajikistan have one genotype each. Only one genotype in this population has an unknown origin. The above-mentioned results from the population structure also confirmed the recorded Nei’s genetic distance among different cotton genotypes. For instance, two genotypes with the highest genetic distance, G42 (originated from Egypt) and G128 (originated from the USA), were grouped separately in two different populations (Pop A and Pop C, respectively). The contrary was also observed in two populations with the lowest genetic distance, G125 (originated from the USA) and G126 (originated from Turkey), where they were both grouped into one sole population C. This may be explained by historic facts dating back to the first half of the twentieth century when it was reported that more than 40 upland cotton (G. hirsutum L.) genotypes from the USA were introduced to Turkey, mainly in two cotton research centers at that time, including Nazilli Cotton Research Institutes, and they were used in several breeding studies intended to improve cotton in Turkey [17]. The exploration of genetic relationships between the studied 128 cotton accessions using neighbor-joining clustering resulted in a comparable clustering pattern to that of model-based algorithm with few exceptions as pure lines were not clustered to each other and accession having high genetic distance were clustered in same group but far each other. The neighbor-joining clustering grouped the studied germplasm into two population i.e. A and B. A total of 28 and 100 genotypes clustered in population A and B, respectively. Population A was further divided into subpopulation A1 and A2 comprising 7 and 21 genotypes respectively. Population comprised genotypes genotypesonly from Türkiye and USA. Population B was further subdivided into sub-populations B1 and B2 comprising a total of 7 and 93 genotypes respectively. The sub-population B1 comprises pure lines like D-Z-P-2, D-Z-P-15, D-Z-P-17, and four genotypes from Türkiye. The sub-population B2 was divided into B2I and B2II and clustered a total of 93 genotyes. It was observed that genotypes from Türkiye and USA were present in both groups by showing their genetic similarity with the genotypes from other countries. One of the possible reason might be seed mixing during collection and conservation at the USDA gene bank. The discrepancies displayed in neighbor-joining clustering might be described by its reduced resolution power relative to the model-based Structure [53]. Principal coordinate analysis (PCoA) confirmed the clustering based on the structure algorithm of 128 cotton accessions into clearly distinguishable five main populations using 11 iPBS-retrotransposons primers (Fig. 5). The occurrence of some light differences between model-based structure and PCoA can derive from its differing clustering resolution, with more resolution revealed by the model-based structure analysis. These differences in clustering were inline with a previous study by Nadeem [54] in which he used the same marker system as to this study in wheat germplasm and also give preference to strcucture based clustering compared to other algortihms.
Conclusion
The iPBS retrotransposon-based marker system is an effective DNA fingerprinting technique for assessing the genetic variation among various crops. This was the first study to elucidate genetic variation and population structure of cotton germplasm using iPBS retrotransposon-based markers. Genotypes G42 and G128 showed high genetic distance and these genotypes might be recommended for future breeding programs. Results of AMOVA explored higher genetic variations within populations compared to among the populations. The model-based structure algorithm and PCoA separated the germplasm into five distinct populations and the highest mean values of diversity indices showed the robustness, applicability and effectiveness of iPBS markers for the assessment of genetic diversity among various crops. This study will be helpful for cotton breeders and researchers for further breeding research on cotton.
Data Availability
The datasets used for the current study ıs provided in this manuscript.
References
Khan MA, Wahid A, Ahmad M, Tahir MT, Ahmed M, Ahmad S, Hasanuzzaman M (2020) World Cotton Production and Consumption: an overview. In: Ahmad S, Hasanuzzaman M (eds) Cotton Production and uses. Springer, Singapore. https://doi.org/10.1007/978-981-15-1472-2_1
Xiao G, Zhao P, Zhang Y (2019) A pivotal role of hormones in regulating cotton fiber development. Front Plant Sci 10(87). https://doi.org/10.3389/fpls.2019.00087
Tyagi P, Gore MA, Bowman DT, Campbell BT, Udall JA, Kuraparthy V (2014) Genetic diversity and population structure in the US Upland cotton (Gossypium hirsutum L). Theor Appl Genet 127(2):283–295. https://doi.org/10.1007/s00122-013-2217-3
Jabran K, Ul-Allah S, Chauhan BS, Bakhsh A (2019) An Introduction to Global Production Trends and Uses, History and Evolution, and Genetic and Biotechnological Improvements in cotton. Hoboken, NJ: wiley online library. https://doi.org/10.1002/9781119385523.ch1
Saleem MA, Qayyum A, Malik W, Amjid MW (2020) Molecular breeding of cotton for Drought stress tolerance. In: Ahmad S, Hasanuzzaman M (eds) Cotton Production and uses. Springer, Singapore. https://doi.org/10.1007/978-981-15-1472-2_1
Basal H, Karademir E, Goren HK, Sezener V, Dogan MN, Gencsoylu I, Erdogan O (2019) Cotton production in turkey and Europe. In: Jabran K, Chauhan BS (eds) Cotton Production. John Wiley & Son Inc., New York, NY, USA, pp 297–321
Hayat K, Bardak A, Parlak D, Ashraf F, Imran HM, Haq HA, Mian MA, Mehmood Z, Akhtar MN (2020) Biotechnology for Cotton Improvement. In: Ahmad S, Hasanuzzaman M (eds) Cotton Production and uses. Springer, Singapore. https://doi.org/10.1007/978-981-15-1472-2_1.
Malik W, Ashraf J, Iqbal MZ, Khan AA, Qayyum A, Abid MA, Noor E, Ahmad MQ, Abbasi GH (2014) Molecular markers and cotton genetic improvement: Current status and future prospects. The Scientific World Journal, 2014, 1–15. https://doi.org/10.1155/2014/607091
Tokel D, Dogan I, Hocaoglu-Ozyigit A, Ozyigit II (2022) Cotton Agriculture in Turkey and Worldwide Economic Impacts of turkish cotton. J Nat Fib 6:1–20
USDA, United States Department of Agriculture Foreign Agricultural Service (2020a), December 2020 Report, Cotton: World Markets and Trade, 1–28. https://downloads.usda.library.cornell.edu/usda-esmis /
Yilmaz I, Akcaoz H, Ozkan B (2005) An analysis of energy use and input costs for cotton production in Turkey. Renewable Energy 30:145–155. https://doi.org/10.1016/j.renene.2004.06.001
Daǧdelen N, Başal H, Yilmaz E, Gürbüz T, Akçay S (2009) Different drip irrigation regimes affect cotton yield, water use efficiency and fiber quality in western Turkey. Agric Water Manage 96(1):111–120. https://doi.org/10.1016/j.agwat.2008.07.003
Rana MK, Bhat KV (2005) RAPD markers for genetic diversity study among indian cotton cultivars. Curr Sci 88(12):1956–1961
Dongre AB, Bhandarkar M, Banerjee S (2007) Genetic diversity in tetraploid and diploid cotton (Gossypium spp.) using ISSR and microsatellite DNA markers. Indian J Biotechnol 6:349–353
Li Z, Wang X, Zhang Y, Zhang G, Wu L, Chi J, Ma Z (2008) Assessment of genetic diversity in glandless cotton germplasm resources by using agronomic traits and molecular markers. Front Agric China 2(3):245–252. https://doi.org/10.1007/s11703-008-0063-x
Murtaza N (2006) Cotton genetic diversity study by AFLP markers. Electron J Biotechnol 9(4):1–5. https://doi.org/10.2225/vol9-issue4-fulltext-9
Hocaoglu-ozyigit A, Ucar B, Altay V, Ozyigit II (2020) Genetic diversity and phylogenetic analyses of turkish cotton (Gossypium hirsutum L.) lines using ISSR markers and Chloroplast trnL-F regions. J Nat Fibers 1–14. https://doi.org/10.1080/15440478.2020.1788493
Nadeem MA, Nawaz MA, Shahid MQ, Doğan Y, Comertpay G, Yıldız M, Hatipoğlu R, Ahmad F, Alsaleh A, Labhane N, Özkan H, Chung G, Baloch FS (2018) DNA molecular markers in plant breeding: current status and recent advancements in genomic selection and genome editing. Biotechnol Biotechnol Equip 32(2):261–285. https://doi.org/10.1080/13102818.2017.1400401
Baloch FS, Alsaleh A, de Miera LES, Hatipoğlu R, Çifti V et al (2015) DNA based iPBS-retrotransposon markers for investigating the population structure of pea (Pisum sativum) germplasm from Turkey. Biochem Syst Ecol 61:244–252. https://doi.org/10.1016/j.bse.2015.06.017
Yıldız M, Koçak M, Baloch FS (2015) Genetic bottlenecks in turkish okra germplasm and utility of iPBS retrotransposon markers for genetic diversity assessment. Genet Mol Res 14:10588–10602. https://doi.org/10.4238/2015.September.8.20
Demirel U, Tındaş İ, Yavuz C, Baloch F, Çalışkan M (2018) Assessing genetic diversity of potato genotypes using inter-PBS retrotransposon marker system. Plant Genetic Resources: Characterization and Utilization 16(2):137–145. https://doi.org/10.1017/S1479262117000041
Yaldız G, Camlica M, Nadeem MA, Nawaz MA, Baloch FS (2018) Genetic diversity assessment in Nicotiana tabacum L. with iPBS-retrotransposons. Turk J Agric For 42:154–164. https://doi.org/10.3906/tar-1708-32
Ali F, Yılmaz A, Nadeem MA, Habyarimana E, Subaşı I et al (2019) Mobile genomic element diversity in world collection of safower (Carthamus tinctorius L.) panel using iPBS-retrotransposon markers. PLoS ONE 14(2):1–19. https://doi.org/10.1371/journal.pone.0211985
Yıldız M, Koçak M, Nadeem MA, Cavagnaro P, Barboza K et al (2019) Genetic diversity analysis in the turkish pepper germplasm using iPBS retrotransposon-based markers. Turk J Agric For 44:1–14. https://doi.org/10.3906/tar-1902-10
Barut M, Nadeem MA, Karaköy T, Baloch FS (2020) DNA fngerprinting and genetic diversity analysis of world quinoa germplasm using iPBS-retrotransposon marker system. Turk J Agric For 44:479–491. https://doi.org/10.3906/tar-2001-10
Shimira F, Boyaci HF, Yeter Ç, Nadeem MA, Baloch FS, Taskin H (2021) Exploring the genetic diversity and population structure of scarlet eggplant germplasm from Rwanda through iPBS retrotransposon markers. Mol Biol Rep. https://doi.org/10.1007/s11033-021-06626-0
Baloch FS, Guizado SJ, Altaf MT, Yüce I, Çilesiz Y, Bedir M, Nadeem MA, Hatipoglu R, Gómez JC (2022) Applicability of inter-primer binding site iPBS-retrotransposon marker system for the assessment of genetic diversity and population structure of peruvian rosewood (Aniba rosaeodora Ducke) germplasm. Mol Biol Rep 49(4):2553–2564
Kalendar R, Antonius K, Smýkal P, Schulman HA (2010) iPBS: a universal method for DNA fingerprinting and retrotransposon isolation. Theor Appl Genet 121:1419–1430. https://doi.org/10.1007/s00122-010-1398-2
Milovanov A, Zvyagin A, Daniyarov A et al (2019) Genetic analysis of the grapevine genotypes of the russian Vitis ampelographic collection using iPBS markers. Genetica 147:91–101. https://doi.org/10.1007/s10709-019-00055-5
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Baloch, F. S., Guizado, S. J. V., Altaf, M. T., Yüce, I., Çilesiz, Y., Bedir, M.,… Gómez, J. C. C. (2022). Applicability of inter-primer binding site iPBS-retrotransposon marker system for the assessment of genetic diversity and population structure of Peruvian rosewood (Aniba rosaeodora Ducke) germplasm. Molecular Biology Reports, 49(4),2553–2564
Yeh FC, Yang R, Boyle TJ, Ye Z, Xiyan JM (2000) PopGene32, Microsoft Windows-based freeware for population genetic analysis, version 1.32. Molecular Biology and Biotechnology Centre—University of Alberta, Edmonton
Roldán-Ruiz I, Dendauw J, Van Bockstaele E, Depicker A, De Loose M (2000) AFLP markers reveal high polymorphic rates in ryegrasses (Lolium spp). Mol Breed 6(2):125–134. https://doi.org/10.1023/A:1009680614564
Peakall ROD, Smouse PE (2006) GENALEX 6: genetic analysis in excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14(8):2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Adawy SS, Hussein EHA, El-itriby HA (2006) Molecular characterization and genetic relationships among cotton genotypes 2- AFLP analysis. Arab J Biotechnol 9(3):477–492
Esmail RM, Zhang JF, Adel-Hamid AM (2008) Genetic diversity in Elite Cotton Germplasm Lines using field performance and rapd markers. World J Agricultural Sci 4(3):369–375
Liu Z, Zhang Y, Zhang S, Deng K, Dong S, Ren Z (2011) Genetic diversity analysis of various red spider mite- resistant upland cotton cultivars based on RAPD. Afr J Biotechnol 10(18):3515–3520. https://doi.org/10.5897/AJB10.832
Noormohammadi Z, Shojaei-jesvaghani F, Sheidai M, Farahani F, Alishah O (2011) Inter simple sequence repeats (ISSR) and random amplified polymorphic DNA (RAPD) analyses of genetic diversity in Mehr cotton cultivar and its crossing progenies. Afr J Biotechnol 10(56):11839–11847. https://doi.org/10.5897/AJB11.1377
Noormohammadi Z, Farahani YH, Sheidai M, Ghasemzadeh-Baraki S, Alishah O (2013a) Genetic diversity analysis in Opal cotton hybrids based on SSR, ISSR, and RAPD markers. Genet Mol Res 12(1):256–269. https://doi.org/10.4238/2013.January.30.12
Zhang Y, Wang XF, Li ZK, Zhang GY, Ma ZY (2011) Assessing genetic diversity of cotton cultivars using genomic and newly developed expressed sequence tag-derived microsatellite markers. Genet Mol Res 10(3):1462–1470. https://doi.org/10.4238/vol10-3gmr1277
De Magalhães Bertini CHC, Schuster I, Sediyama T, de Barros EG, Moreira MA (2006) Characterization and genetic diversity analysis of cotton cultivars using microsatellites. Genet Mol Biology 29(2):321–329. https://doi.org/10.1590/S1415-47572006000200021
Chen G, Du, Xiong-ming D (2006) Genetic diversity of source germplasm of Upland Cotton in China as determined by SSR marker analysis. Acta Genetica Sinica 33(8):733–745. https://doi.org/10.1016/S0379-4172(06)60106-6
Khan AI, Fu Y, Khan IA (2009) Genetic diversity of pakistani cotton cultivars as revealed by simple sequence repeat markers. Commun Biometry Crop Sci 4(1):21–30
Campbell BT, Williams VE, Park W (2009) Using molecular markers and w eld performance data to characterize the Pee Dee cotton germplasm resources. Euphytica 169:285–301. https://doi.org/10.1007/s10681-009-9917-4
Tu JL, Zhang MJ, Wang XQ, Zhang XL, Lin ZX (2014) Genetic dissection of upland cotton (Gossypium hirsutum) cultivars developed in Hubei Province by mapped SSRs. Genet Mol Res 13(1):782–790. https://doi.org/10.4238/2014.January.31.4
Ge H, Liu Y, Jiang M, Zhang J, Hana H et al (2013) Analysis of genetic diversity and structure of eggplant populations (Solanum melongena L.) in China using simple sequence repeat markers. Sci Hortic 162:71–75. https://doi.org/10.1016/j.scienta.2013.08.004
De Menezes IPP, Hoffmann LV, Barroso APV (2015) Genetic characterization of cotton landraces found in the Paraíba and Rio Grande do norte states. Crop Breed Appl Biotechnol 15:26–32. https://doi.org/10.1590/1984-70332015v15n1a4
Noormohammadi Z, Rahnama A, Sheidai M (2013b) EST-SSR and SSR analyses of genetic diversity in diploid cotton genotypes from Iran. Nucleus 56(3):171–178. https://doi.org/10.1007/s13237-013-0094-4
Yildiz E, Sümbül A, Yaman M, Nadeem MA, Say A, Baloch FS, Popescu GC (2022) Assessing the genetic diversity in hawthorn (Crataegus spp.) genotypes using morphological, phytochemical and molecular markers. Genetic Resources and Crop Evolution, pp 1–12
Baloch FS, Guizado SJV, Altaf MT, Yüce I, Çilesiz Y, Bedir M, Gómez JCC (2022) Applicability of inter-primer binding site iPBS-retrotransposon marker system for the assessment of genetic diversity and population structure of peruvian rosewood (Aniba rosaeodora Ducke) germplasm. Mol Biol Rep 49(4):2553–2564
Alsaleh A, Bektas H, Baloch FS, Nadeem MA, Özkan H (2022) Turkish durum wheat conserved ex-situ and in situ unveils a new hotspot of unexplored genetic diversity. Crop Sci 62(3):1200–1212
Bouchet S, Pot D, Deu M, Rami JF, Billot C, Perrier X et al (2012) Genetic structure, linkage disequilibrium and signature of selection in sorghum: lessons from physically anchored DArT markers. PLoS ONE 7:3
Nadeem MA (2021) Deciphering the genetic diversity and population structure of turkish bread wheat germplasm using iPBS-retrotransposons markers. Mol Biol Rep 48(10):6739–6748
Acknowledgements
We would like to thank the Nazilli Cotton Research Institute and Prof. Dr. Mefhar Gültekin TEMİZ from the Faculty of Agriculture, University of Dicle (Diyarbakir, Turkey) for their assistance in supplying the cotton genetic materials used in this study.
Funding
Authors are grateful to Dicle University - Scientific Research Projects Coordinating Office for financial support (Project No: ZİRAAT.20.004).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of interest
The authors declare no conflicts of interests.
Consent to publish
All authors read the manuscript and showed their willingness to publish this study.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Baran, N., Shimira, F., nadeem, M.A. et al. Exploring the genetic diversity and population structure of upland cotton germplasm by iPBS-retrotransposons markers. Mol Biol Rep 50, 4799–4811 (2023). https://doi.org/10.1007/s11033-023-08399-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-023-08399-0