
Abstract
Patients with β-thalassemia suffer from a lack or absence of the beta-globin chain of normal hemoglobin (Hb). Therefore, an increase in fetal Hb (HbF) levels could improve the clinical status of these patients. Downregulation of BCL11A, a key regulatory transcription factor, could ameliorate the clinical status of thalassemic patients by increasing HbF levels. miR-30a expression and its relationship with the BCL11A gene in erythroid precursors was explored in patients with β-thalassemia. The relevance of miR-30a to clinical parameters was also investigated. We evaluated the expressions of miR-30a, BCL11A, and γ-globin genes by quantitative real-time PCR (qRT-PCR) on isolated erythroid precursors from peripheral blood samples of β-thalassemia intermedia (TI) patients and in bone marrow samples from healthy individuals as controls. The correlation between miR-30a expression and clinical indices that included HbF levels, ferritin, and the frequency of blood transfusions were assessed. We observed increased expression of miR-30a in conjunction with decreased BCL11A expression and elevated γ-globin and HbF levels. Patients with elevated miR-30a expression had a higher percentage of HbF and a lower level of ferritin. In addition, we observed that overexpression of miR-30a in erythroid precursor cells led to reduced BCL11A expression and was associated with elevated γ-globin expression. Our findings showed the importance of miR-30a in BCL11A and HbF regulation, and in the clinical status of patients with β-thalassemia
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
β-thalassemia is a hereditary blood disorder characterized by decreased or lack of synthesis of beta-globin chains, which result in excessive α-chains in red blood cells (RBCs) and ineffective erythropoiesis [1]. Symptomatic cases occur in one out of every 100 000 individuals on average. A higher prevalence of thalassemia has been reported in Africa, central Asia, the Middle East, and the Mediterranean [2]. The severity of thalassemia varies depending on the loss of one or both beta-globin genes. According to its symptoms, thalassemia is classified into three forms—minor, intermediate, and major. Although patients with thalassemia intermedia (TI) frequently lead normal lives, they might require regular transfusions based on the severity of their anemia [3]. Hemoglobin (Hb) levels in TI patients are generally between 7–10 g/dL and symptoms of anemia may not be observed until middle age. These patients are categorized into two distinct classes, transfusion-dependent thalassemia (TDT) and non-transfusion-dependent thalassemia (NTDT). Previous studies have reported the development of iron overload in both groups [4]. Depending on the number of transfusions per year, patients are categorized as chronically transfused (8 ≤ transfusions), intermittently transfused (1–7 transfusions), or never transfused [5]. Elevations in fetal Hb (HbF) levels lead to compensation by ineffective erythropoiesis, which prevents anemia and transfusions, and, in turn, iron overload [6]. Genome-wide association studies (GWAS) can detect susceptible loci that are responsible for controlling HbF levels. These genes include HBS1L/MYB, B cell CLL/lymphoma 11A (BCL11A), and β-like globin gene cluster, each of which might demonstrate significant heterogeneity and different frequencies of alleles [7]. In addition to these genetic factors, gene polymorphism could influence the levels of HbF and F cells. The Xmn1 polymorphism (C → T 158) is a recurrent variant of the γ-globin gene, which accounts for almost 30% of the variations [8, 9]. The presence of the Xmn1(G) polymorphism with an IVS-II-1 mutation reportedly leads to delayed clinical manifestations and affects therapy progress with hydroxyurea [10].
In addition to HbF inducer drugs and gene polymorphism, naturally occurring non-coding RNAs (ncRNA), which are called microRNAs (miRNAs), also effectively regulate gene expression and play a crucial role in globin gene expression and reactivation of γ-globin [11]. Several miRNAs are associated with developmental changes in globin gene expression and reactivation of γ-globin. While adults can maintain variable levels of HbF without any adverse clinical outcomes, relatively high levels of HbF can help patients who suffer from Hb disorders such as β-thalassemia [12]. miR-210, miR-221, miR-222, miR-15a, miR-16, mir-34a, miR-96, miR-486-3p, let-7, miR-326, and miR-144 function as regulators of HbF in erythroid cells via modulating factors such as BCL11A, stem cell factor (SCF), MYB, NRF2, and EKLF [13,14,15,16,17,18,19,20,21]. BCL11A is a zinc finger protein that is necessary for normal development of lymphocytes and erythroid lineage. BCL11A expresses in hematopoietic progenitor cells (HPCs) and is downregulated during myeloid differentiation [22,23,24]. The results of recent genetic studies have found that BCL11A is a new regulator for silencing γ-globin expression in erythroid precursors and a developmental regulator of Hb switching. It has been shown that mediated knockdown of BCL11A by a lentiviral vector in erythroid cells was associated with increased levels of HbF production, with no changes in erythroid differentiation. This indicates that BCL11A regulates HbF levels through direct transcriptional inhibition of the γ-globin gene, rather than via modulation of erythroid kinetics, as suggested for the HBS1L-MYB locus [13]. miR-30a is located on chromosome 6q.13 and reportedly prevents tumor metastasis and proliferation, control of epithelial to mesenchymal transition (EMT), and plays a role in chronic myeloid leukemia (CML) autophagy [25]. The results of a cohort study on patients with non-small cell lung cancer (NSCLC) have shown a remarkable relation between disease pathogenesis and miR-30a expression as a suppressor of BCL11A [26, 27]. Based on the important role of BCL11A on Hb switching and inhibiting this factor by miR-30a in previous studies, in the present study, we investigated the underlying mechanism for the roles of miR-30a and BCL11A in governing γ-globin gene expression and assessed the correlation between miR-30a and HbF levels. We found that miR-30a suppressed BCL11A expression and increased γ-globin gene expression. A higher expression of miR-30a was related to an increased level of HbF in TI patients.
Materials and methods
Sample collection and laboratory analysis
This study was conducted on samples provided by patients diagnosed with TI (n = 31). Human adult peripheral blood and bone marrow samples (n = 3) were procured and processed following approval by the Ethics Committee of Tarbiat Modares University, Tehran, Iran (registration no. 2398007, approved September 02, 2016) and after patients provided written informed consent for study participation. Peripheral blood (5–10 mL) was collected at the Zafar Adult Thalassemia Clinic, Iranian Blood Transfusion Organization (IBTO), Tehran, Iran during 2017–2018. None of the patients were under treatment with hydroxyurea, nor had they received any transfusions for at least the past month prior to sample collection. The blood samples were collected in EDTA tubes and analyzed by an automated hematology analyzer (Sysmex KX-21N, Japan). Serum ferritin was analyzed using an automated immunoassay system (Architect i1000SR; Abbott Diagnostics, USA) to evaluate iron overload status. HbF also was measured using a capillary electrophoresis system (Sebia Capillarys 2 Flex Piercing system, France).
Molecular analysis
DNA was extracted from the patients’ peripheral blood leukocytes by the salting out method. The most common mutations were genotyped by PCR in an Eppendorf, Mastercycler® gradient 5331 (Eppendorf AG, Hamburg, Germany). ARMS-PCR and beta-globin gene sequencing were performed to detect β-thalassemia gene mutations. In the case of non-detecting expected mutations, α-globin gene mutations were assessed using GAP-PCR (3.7, 4.2, and MED) in addition to HBA1 and HBA2 gene DNA sequencing. We used RLFP-PCR to detect the Xmn1 polymorphism in patients’ samples. Blank solutions that contained all of the components, except for DNA, were tested with the samples to prevent any false positives. All reactions were performed in triplicate.
Enrichment of erythroid precursor cells
Erythroid precursors were isolated from 2 mL of normal bone marrow and 4 ml peripheral blood from the patients diagnosed with TI using Percoll (Sigma-Aldrich, USA) as previously described [28].
Isolation of hematopoietic stem cells (HSCs)
Hematopoietic stem cells)HSCs( were isolated from the buffy coat derived from 150 mL of donated cord blood (CB). Preservative-free CB samples that contained citrate phosphate dextrose (CPD) anti-coagulant were collected from the IBTO Public Umbilical Cord Blood Center. Red cells were initially precipitated using 6% hydroxyl-ethyl- starch (HES, Spain). Mononuclear cells (MNCs) were isolated from the supernatant by density gradient centrifugation on Ficoll-Hypaque (Lymphodex, Germany) as previously described [29]. Next, the CD34+ HPCs were purified with a CD34 Microbead Kit according to the manufacturer’s instructions (Miltenyi Biotec, Germany).
Cell culture and nucleofection
For erythroid differentiation, the isolated CD34+ cells were cultivated in semi-solid media (Methocult; STEMCELL Technologies, Canada) that contained 2 U/mL erythropoietin (EPO), 10 ng/mL IL-3, and 100 ng/mL SCF (all from PeproTech, USA). The cells were maintained for 10 days at 37 °C in a humidified atmosphere and 5% CO2. At day 10, burst-forming unit-erythroid (BFU-E) colonies that consisted of pure erythroid precursors were harvested by using a Pasteur pipette and a stereomicroscope, under a class I biological safety cabinet. The colonies were washed twice with ice-cold phosphate-buffered saline (PBS; Gibco, USA), and the erythroid precursors (2 × 106 cells) were centrifuged at 200×g for 10 min at room temperature, then re-suspended in 100 µL human CD34 Cell Nucleofector™ Solution (Amaxa GmbH, Köln, Germany). The cell suspension was combined with 5 μg of a LentimiRa-GFP-hsa-miR-30a-5p plasmid (ABM, Canada). The cell/DNA suspension was transferred to a certified cuvette and transfected using program U-008 of the nucleofector device (Amaxa GmbH). The negative control consisted of mock transfected cells, which were nucleofected with plasmid that lacked precursor-miR-30a. After nucleofection, 500 μL pre-warmed culture medium was added to the cuvette that contained the nucleofected cells and the cells were transferred to 12-well plates. The cells were incubated at 37 °C for 48 h. miR-30a expression was analyzed by qPCR to assess the transfection efficiency.
CD34 purity and erythroid lineage differentiation
Flow cytometry was performed to assess the purity of the isolated CD34+ HPSCs by using PE-conjugated human CD34 antibody (BD Pharmingen, USA). The purity of the isolated erythroid precursor cells and the CD34+ differentiated erythroid lineage was measured using anti-CD235a-FITC-conjugated (BD Pharmingen, USA) and anti-CD71-PE-conjugated (BD Pharmingen) antibodies according to previously described protocols [30]. Finally, the cells were washed twice with PBS and resuspended in 100 μL PBS that contained 0.5% BSA. Flow cytometry analysis was performed within an hour after the preparation procedure with a FACSCalibur instrument (Becton Dickinson, USA). Data analyses were carried out using FlowJo 7.5.5 software.
RNA extraction and q-RT PCR
Total cellular RNA that included miRNAs was extracted from at least 2 × 105 cells for each sample by using TRIzol reagent (Thermo Fisher Scientific, USA) following the manufacturer’s instructions. The concentration and purity of the RNA samples was assessed as 260/280 nm and 260/230 nm ratios, respectively, and were measured by a BioPhotometer 6131 (Eppendorf, Germany). cDNA was synthesized with a PrimeScript™ RT-PCR kit according to the manufacturer’s instructions (Takara Bio, Inc., Japan). cDNA synthesis for evaluation of miR-30a was performed by a different procedure using a Pars Genome miRNA Assay kit according to the protocol (Pars Genome, Iran). The following primers were used for qPCR: BCL11A sense: 5′-ACA GGA ACA CAT AGC AGA TAA AC-3′ and antisense: 5′-TAT TCT GCA CTC ATC CCA GG-3′; γ-globin sense: 5′-CTG GGA AGG CTC CTG GTT G-3 and antisense: 5′-CAG AGG CAG AGG ACA GGT TG-3′; β-actin sense: 5′-TGA AGA TCA AGA TCA TTG CTC CTC-3′ and antisense: 5′-AGT CAT AGT CCG CCT AGA AGC-3′. Quantitative real-time PCR (qRT-PCR) was performed using the StepOne Real-time PCR System (Applied Biosystems, USA). RNU6 was the internal control in miRNA quantification. The PCR efficiency for all of the analyzed genes was estimated using standard curves generated by PCR on serial dilutions of cDNA.
Statistical analysis
The normality of the sample distributions was calculated by the Kolmogorov-Smirnoff test. Because the samples had normal distribution, we used the t-test and one-way ANOVA to assess the data differences between the TI patients. Correlation between miR‑30a and HbF was assessed by the Pearson's correlation test. Statistical analysis was carried out with GraphPad Prism, version 6.07 (GraphPad Software, USA). A p-value < 0.05 was considered to be statistically significant.
Results
Genetic assessment of patients with thalassemia intermedia (TI)
All patients (n = 31) were confirmed to have typical β-thalassemia mutations (Table 1), which have the highest incidence among Iranian thalassemia patients. The IVS-II-1 mutation was the dominant mutation among the mutations in the studied thalassemia samples. No patients had any α-globin gene mutations (Table 1). Patients were also evaluated for the presence of the Xmn1 polymorphism. The results indicated that 61% of patients had this polymorphism. Carriers of the Xmn1 polymorphism have a higher level of HbF and a better clinical manifestation. As shown in Fig. 1a, a significant relationship existed between the level of HbF in the homozygous and wild type (no polymorphism) groups (p < 0.001). There was no significant correlation noted between miR-30a expression and the Xmn1 polymorphism (Fig. 1b).

The Xmn1 polymorphism and its relationship with fetal hemoglobin (HbF; %) and miR-30a expression in patients with thalassemia intermedia (TI) (n = 31). a A significant relationship existed between HbF (%) in the group without any polymorphism and the homozygous group. b Gene expression analysis of miR-30a in the group without any polymorphism, and the heterozygous and homozygous groups by qRT-PCR. Data are presented as SD. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001
Fetal hemoglobin (HbF) content and flow cytometry assay
HbF level was measured by capillary electrophoresis. All patients with TI had an increased level of HbF (average: 57.67%) (Table 1). Flow cytometry histogram showed that the purity of the CD34+ cells was 98% (Fig. 2a). A total of 76.1% of the differentiated erythroid precursors expressed CD71 and 97.6% expressed CD235a (Fig. 2b, c).

The CD34+ hematopoietic stem cells (HSCs) and erythroid precursors were characterized by analyzing the CD34, CD71, and CD235a markers, respectively. a A total of 98% of isolated cells expressed CD34. b, c Erythroid precursors were expressed in both the CD71 and CD235a markers
Increased miR-30a expression in thalassemic erythroid precursors
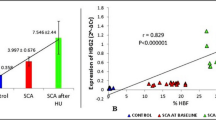
We assessed expressions of miR-30a, BCL11A, and γ-globin in the erythroid precursors of normal bone marrow and patients with TI. The results showed a significantly higher miR-30a level in patients with TI compared with the normal samples (p < 0.0001; Fig. 3a). Additionally, an 11.6-fold increase in miR-30a was coupled with a 0.62-fold decrease in BCL11A (p < 0.05; Fig. 3b). γ-globin gene expression increased by 12.26-fold in patients with TI compared with the normal group (p < 0.001; Fig. 3c). We determined the correlation between miR-30a and HbF levels to better assess the relative abundance of miR-30a expression. The miR-30a fold changes had a positive correlation with HbF levels (r = 0.8004, p < 0.0001) in erythroid precursors from patients with TI.

miR-30a affected BCL11A and γ-globin gene expressions. a–c Expression levels of miR-30a, BCL11A, and γ-globin genes in erythroid precursors of thalassemia intermedia (TI) and normal bone marrow aspiration by quantitative real-time PCR (qRT-PCR). d A meaningful correlation existed between the levels of miR-30a and fetal hemoglobin (HbF). Expressions of γ-globin and BCL11A were normalized with the β-actin and miR-30a genes by RNU6. Data are depicted as SD. **p < 0.01, ***p < 0.001, and ****p < 0.0001
miR-30a expression and status of patients’ transfusions
Correlation between the level of miR-30a and transfusion status was examined to evaluate the effect of miR-30a levels on transfusion and non-transfusion dependent patients with TI. Approximately 39% of the patients (n = 12) had never received transfusions and 61% (n = 19) were intermittently transfused (Fig. 4a). There was a higher level of miR-30a in patients who had never been transfused (p < 0.0009, Fig. 4b). A comparison of HbF levels between both groups showed a significantly higher level in patients with no transfusions (66.9%) compared with the intermittently transfused group (44.77%; p = 0.04) (Fig. 4c). We measured serum ferritin levels to evaluate the link between miR-30a and iron overload. The ferritin level was significantly lower in the group with no transfusions compared with the intermittent group. On the other hand, a lower ferritin level (790 ng/ml) was associated with a higher level of miR-30a (456 ng/ml) (Fig. 4d).

Parameters such as fetal hemoglobin (HbF), ferritin, and miR-30a gene expressions in relation to the transfusion status of patients with thalassemia. a The numbers of patients with thalassemia intermedia (TI) in the intermittent and never-transfused groups. b miR-30a gene expression was significantly higher in the intermittent group compared with the never-transfused patients with TI. c Comparison of HbF level in never-transfused and intermittently-transfused patients showed that the never-transfused patients had a higher percentage of HbF. d The never-transfused group had a lower ferritin level. Data are presented as SD. ***p < 0.001
Overexpression of miR-30a and BCL11A level
miR-30a expression increased significantly after transfection of the erythroid precursor cells within 24 h (4.5-fold) and by 7.18-fold within 48 h compared to the control group (Fig. 5a, d; p < 0.05). The cells transfected with miR-30a vector had decreased BCL11A expression within 24 and 48 h compared to the control group; however, this decrease was only significant at 48 h (Fig. 5b, e; p < 0.05). γ-globin gene expression increased in cells transfected with miR-30a by 1.49-fold at 24 h and by 6.87-fold at 48 h (Fig. 5c, f; p < 0.05).

Evaluation of miR-30a over-expression on the studied genes, including BCL11A and γ–globin. a, d miR-30a expression increased significantly in erythroid precursors after transfection with the precursor-hsa-miR-30a plasmid. b–e Significant downregulation of BCL11A expression in the transfected miR-30a group occurred after 48 h of transfection. d–f γ-globin gene expression increased significantly in the miR-30a transfected group after 48 h compared with the scrambled group. *p < 0.05, **p < 0.01
Discussion
Thalassemia is one of the most common types of anemia associated with β-globin defects. Major and intermediate thalassemia patients suffer from problems caused by anemia. One of the principal treatments to alleviate symptoms is to increase HbF levels by targeting genes responsible for the switch from HbF to HbA and by inhibition of BCL11A gene expression [6]. miRNAs are novel effector molecules that can target γ-globin negative regulatory genes to alter HbF levels [31]. Many studies have investigated the role of miRNA and HbF induction in hemoglobinopathies. The results of in vitro studies have demonstrated that miR-96 directly targets the ORF of γ-globin to mediate gene silencing [15]. Furthermore, individuals with trisomy 13 have elevated HbF levels due to inhibition of the repressor protein MYB by miR-15a and miR-16 [32]. A previous study in patients with β-thalassemia has shown an association between elevated miR-486-3p and higher HbF levels linked to reduced expression of BCL11A, a developmentally regulated repressor of γ-globin [13]. These studies, among others, indicate the role of miRNAs as prognostic, diagnostic or even clinical targets for β-thalassemia treatment. Hence, these observations have prompted us to determine whether miR-30a expression is subject to miRNA regulation in human erythroid cells. A significant relationship has been identified between elevated miR-30a expression and its association with BCL11A in NSCLC. In this study, pmirGLO dual-luciferase reporter plasmids that carry the 3′-UTR sequence of BCL11A and plasmids that expressed miR-30a, miR-1 or a control miRNA were co-transfected into the A549 and NIH3T3 cell lines. Significant repression was seen only in miR-30a-transfected cells. As a result, it was reported that inactivation of miR-30a might contribute to proto-oncogene BCL11A activation [26]. Since it was confirmed that miR-30a targets BCL11A, as a master regulator of γ-globin expression, we were interested in the potential association between miR-30a, BCL11A, and γ-globin in erythroid precursors of thalassemia patients.
The present study indicated that the 61% of TI patients had the Xmn1 polymorphism and IVS-II-1 was the most prevalent mutation. The presence of the IVS-II-1 mutation, either with or without Xmn1, was proven to aide indicators for diagnosis of TI [33]. We assessed the relation between the Xmn1 polymorphism and HbF levels and found a significant relationship between HbF levels in patients without the polymorphism and homozygous patients. Our results supported the results of other studies where it was shown that, at most, an 89% HbF level variation in different individuals was regulated by genetic factors, among which the Xmn1 polymorphism had a remarkable role [9]. Researchers in another study reported that this polymorphism was more prevalent in patients with TI and had a strong relationship with the IVS-II-1 mutation [34]. The Xmn1 polymorphism and miR-30a, both of which were noticeably elevated in our patients, potentially increase HbF levels. We sought to determine the cause for this increase in HbF and assess the relationship between the Xmn1 polymorphism and miR-30a. Our assessment of the relationship between the Xmn1 polymorphism and miR-30a was not significant in terms of fold change between the three groups, with and without the polymorphism.
The present study evaluated the possible correlation between miR-30a expression with several factors, including BCL11A and γ-globin expressions, percentage of HbF, and transfusion status in erythroid precursors of patients with TI. BCL11A expression had a negative correlation with miR-30a, whereas both γ-globin and HbF had positive relationships with miR-30a. We observed that patients with elevated miR-30a expression had increased levels of HbF due to the lower levels of BCL11A. The results of a previous study demonstrated that miR-451 upregulation occurred on day 3 of thalassemic peripheral blood CD34+ cell differentiation into erythroid cells. This study also suggested that early erythroid precursors in patients with thalassemia had abnormal miR-451 expression, which could be used to determine abnormalities of erythropoiesis [35]. Siwaponanan et al. reported that miR-210 had an inverse correlation to Hb and Hct. They observed that a higher level of miR-210 in the RBCs of patients with thalassemia might be engendered by hypoxia, which resulted from decreased Hb levels [36].
With respect to the noticeable relationship between miR-30a and increased HbF, we analyzed patients’ clinical conditions such as transfusion status and iron overload to further assess this impact. The increase in HbF had a stable association with an improved patient’s clinical status, which included reduced ineffective erythropoiesis, α-chain accumulation, and iron overload. Increased HbF levels were an effective treatment for patients with thalassemia [37]. HbF levels were increased in both the intermittently-transfused and never-transfused groups. This increase was significantly higher in the never-transfused group compared with the intermittently-transfused group. We observed that the increased miR-30a expression was higher in the never-transfused group. Serum ferritin levels were lower in the never-transfused group. The higher level of miR-30a was consistent with a lower dependence on transfusion. When the HbF level increased, dependence of patients on transfusions decreased, which reduced iron overload [6]. Additionally, we assumed with an increased HbF level, patients would have less hypoxia and anemia. Consequently, hepcidin, as a master regulator of iron absorption, would increase and patients would have lower levels of ferritin and iron overload [38]. Recently, it was found that under hypoxic conditions, high levels of miR-30a were rich in cardiomyocyte exosomes. This study mentioned that miR-30a expression is regulated by HIF-1α, which is efficiently transferred via exosomes between cardiomyocytes after induction of hypoxia [39]. Patients with thalassemia suffer from anemia and, consequently, hypoxia, which causes increased expression of hypoxia transcription factors such as HIF-1α [38]. As a result, anemia and hypoxia directly amplify ineffective erythropoiesis. Our results supported this as evidenced by the higher fold changes of miR-30a in patients with TI.
Increased miR-30a expression, other than HbF, appeared to have a negative correlation with ferritin levels in erythroid precursors in patients with TI compared to normal controls. This finding suggested that miR-30a might play a pivotal role in governing globin switching, induction of HbF, and iron overload status during stressed erythropoiesis in patients with β-thalassemia. We also overexpressed miR-30a in normal erythroid precursors. BCL11A expression diminished following an increase in miR-30a expression with an increase in γ-globin mRNA levels. As a result, it was confirmed that a transient increase in miR-30a expression had a crucial effect on BCL11A and γ-globin expression, where miR-30a and BCL11A were endogenously expressed. Notably, the same scenario was seen in erythroid precursors of patients with TI. A similar study investigated miRNA roles, such as miR-486-3p, in HbF elevation. This miRNA increased γ-globin gene expression and HbF by inhibiting BCL11A [13].
It seems reasonable to presume that miR-30a regulates HbF levels and ameliorate symptoms of anemia. This, in turn, decreases both dyserythropoiesis and the frequency of blood transfusions. We have identified miR-30a as a direct regulator of BCL11A expression. Thus, it is a crucial inducer of HbF by regulation of BCL11A. Analyzing miR-30a in conjunction with other blood indexes can help us to improve our evaluation of the clinical status of patients with thalassemia.
References
Mettananda S, Gibbons RJ, Higgs DR (2015) α-Globin as a molecular target in the treatment of β-thalassemia. Blood J Am Soc Hematol 125(24):3694–3701
Dimou NL, Pantavou KG, Bagos PG (2017) Apolipoprotein E polymorphism and left ventricular failure in beta-thalassemia: a multivariate meta-analysis. Ann Hum Genet 81(5):213–223
Nienhuis AW, Nathan DG (2012) Pathophysiology and clinical manifestations of the β-thalassemias. Cold Spring Harb Perspect Med 2(12):a011726
Voskaridou E, Balassopoulou A, Boutou E, Komninaka V, Christoulas D, Dimopoulou M, Delaki EE, Loukopoulos D, Terpos E (2014) Pregnancy in beta-thalassemia intermedia: 20-year experience of a Greek thalassemia center. Eur J Haematol 93(6):492–499
Vichinsky E, Neumayr L, Trimble S, Giardina PJ, Cohen AR, Coates T, Boudreaux J, Neufeld EJ, Kenney K, Grant A (2014) Transfusion complications in thalassemia patients: a report from the C enters for D isease C ontrol and P revention (CME). Transfusion 54(4):972–981
Sripichai O, Fucharoen S (2016) Fetal hemoglobin regulation in β-thalassemia: heterogeneity, modifiers and therapeutic approaches. Expert Rev Hematol 9(12):1129–1137
Thein SL, Menzel S, Lathrop M, Garner C (2009) Control of fetal hemoglobin: new insights emerging from genomics and clinical implications. Hum Mol Genet 18(R2):R216–R223
Thein SL, Menzel S (2009) Discovering the genetics underlying foetal haemoglobin production in adults. Br J Haematol 145(4):455–467
Khelil AH, Morinière M, Laradi S, Khelif A, Perrin P, Chibani JB, Baklouti F (2011) Xmn I polymorphism associated with concomitant activation of Gγ and Aγ globin gene transcription on a β0-thalassemia chromosome. Blood Cells Mol Dis 46(2):133–138
Said F, Abdel-Salam A (2015) XmnI polymorphism: relation to β-thalassemia phenotype and genotype in Egyptian children. Egypt J Med Hum Genet 16(2):123–127
Choong ML, Yang HH, McNiece I (2007) MicroRNA expression profiling during human cord blood-derived CD34 cell erythropoiesis. Exp Hematol 35(4):551–564
Bauer DE, Kamran SC, Orkin SH (2012) Reawakening fetal hemoglobin: prospects for new therapies for the β-globin disorders. Blood 120(15):2945–2953
Lulli V, Romania P, Morsilli O, Cianciulli P, Gabbianelli M, Testa U, Giuliani A, Marziali G (2013) MicroRNA-486-3p regulates γ-globin expression in human erythroid cells by directly modulating BCL11A. PLoS ONE 8(4):e60436
Gasparello J, Fabbri E, Bianchi N, Breveglieri G, Zuccato C, Borgatti M, Gambari R, Finotti A (2017) BCL11A mRNA targeting by miR-210: a possible network regulating γ-globin gene expression. Int J Mol Sci 18(12):2530
Azzouzi I, Moest H, Winkler J, Fauchère J-C, Gerber AP, Wollscheid B, Stoffel M, Schmugge M, Speer O (2011) MicroRNA-96 directly inhibits γ-globin expression in human erythropoiesis. PLoS ONE 6(7):e22838
Ward CM, Li B, Pace BS (2016) Stable expression of miR-34a mediates fetal hemoglobin induction in K562 cells. Exp Biol Med 241(7):719–729
Pounds CR, Takezaki M, Li B, Ward C, Lopez N, Pace BS (2017) mIR-16 mediated MYB gene silencing induces fetal hemoglobin expression. FASEB J 31(1):979.976
Sankaran VG, Menne TF, Šćepanović D, Vergilio J-A, Ji P, Kim J, Thiru P, Orkin SH, Lander ES, Lodish HF (2011) MicroRNA-15a and-16-1 act via MYB to elevate fetal hemoglobin expression in human trisomy 13. Proc Natl Acad Sci 108(4):1519–1524
Gabbianelli M, Testa U, Morsilli O, Pelosi E, Saulle E, Petrucci E, Castelli G, Giovinazzi S, Mariani G, Fiori ME (2010) Mechanism of human Hb switching: a possible role of the kit receptor/miR 221–222 complex. Haematologica 95(8):1253–1260
Lessard S, Beaudoin M, Orkin SH, Bauer DE, Lettre G (2018) 14q32 and let-7 microRNAs regulate transcriptional networks in fetal and adult human erythroblasts. Hum Mol Genet 27(8):1411–1420
Li Y, Liu D, Zhang X, Li Z, Ye Y, Liu Q, Shen J, Chen Z, Huang H, Liang Y (2018) miR-326 regulates HbF synthesis by targeting EKLF in human erythroid cells. Exp Hematol 63(33–40):e32
Yu Y, Wang J, Khaled W, Burke S, Li P, Chen X, Yang W, Jenkins NA, Copeland NG, Zhang S (2012) Bcl11a is essential for lymphoid development and negatively regulates p53. J Exp Med 209(13):2467–2483
Tsang JC, Yu Y, Burke S, Buettner F, Wang C, Kolodziejczyk AA, Teichmann SA, Lu L, Liu P (2015) Single-cell transcriptomic reconstruction reveals cell cycle and multi-lineage differentiation defects in Bcl11a-deficient hematopoietic stem cells. Genome Biol 16(1):178
Jawaid K, Wahlberg K, Thein SL, Best S (2010) Binding patterns of BCL11A in the globin and GATA1 loci and characterization of the BCL11A fetal hemoglobin locus. Blood Cells Mol Dis 45(2):140–146
Jiang L-h, Zhang H-d, Tang J-h (2018) MiR-30a: A novel biomarker and potential therapeutic target for cancer. J Oncol 2018:5167829
Jiang B-y, Zhang X-c, Su J, Meng W, Yang X-n, Yang J-j, Zhou Q, Chen Z-y, Chen Z-h, Xie Z (2013) BCL11A overexpression predicts survival and relapse in non-small cell lung cancer and is modulated by microRNA-30a and gene amplification. Mol Cancer 12(1):61
Tang R, Liang L, Luo D, Feng Z, Huang Q, He R, Gan T, Yang L, Chen G (2015) Downregulation of MiR-30a is associated with poor prognosis in lung cancer. Med Sci Monit 21:2514
Kwon K, Jeon Y, Hwang H, Lee K, Kim Y, Chung H, Pang M (2007) A high yield of fetal nucleated red blood cells isolated using optimal osmolality and a double-density gradient system. Prenat Diagn 27(13):1245–1250
Delalat B, Pourfathollah AA, Soleimani M, Mozdarani H, Ghaemi SR, Movassaghpour AA, Kaviani S (2009) Isolation and ex vivo expansion of human umbilical cord blood-derived CD34+ stem cells and their cotransplantation with or without mesenchymal stem cells. Hematology 14(3):125–132
Kouhkan F, Hafizi M, Mobarra N, Mossahebi-Mohammadi M, Mohammadi S, Behmanesh M, Zomorrod MS, Alizadeh S, Lahmy R, Daliri M (2014) miRNAs: a new method for erythroid differentiation of hematopoietic stem cells without the presence of growth factors. Appl Biochem Biotechnol 172(4):2055–2069
Saki N, Abroun S, Soleimani M, Kavianpour M, Shahjahani M, Mohammadi-Asl J, Hajizamani S (2016) MicroRNA expression in β-thalassemia and sickle cell disease: a role in the induction of fetal hemoglobin. Cell J (Yakhteh) 17(4):583
Sankaran VG, Menne TF, Šćepanović D, Vergilio J-A, Ji P, Kim J, Thiru P, Orkin SH, Lander ES, Lodish HF (2011) MicroRNA-15a and -16-1 act via MYB to elevate fetal hemoglobin expression in human trisomy 13. Proc Nat Acad Sci U S A 108(4):1519–1524
Ansari S, Rashid N, Hanifa A, Siddiqui S, Kaleem B, Naz A, Perveen K, Hussain Z, Ansari I, Jabbar Q (2018) Laboratory diagnosis for thalassemia intermedia: Are we there yet? J Clin Lab Anal 33:e22647
Miri-Moghaddam E, Bahrami S, Naderi M, Bazi A, Karimipoor M (2017) Xmn1-158 γGvariant in B-thalassemia intermediate patients in South-East of Iran. Int J Hematol Oncol Stem Cell Res 11(2):165
Svasti S, Masaki S, Penglong T, Abe Y, Winichagoon P, Fucharoen S, Umemura T (2010) Expression of microRNA-451 in normal and thalassemic erythropoiesis. Ann Hematol 89(10):953–958
Siwaponanan P, Fucharoen S, Sirankapracha P, Winichagoon P, Umemura T, Svasti S (2016) Elevated levels of miR-210 correlate with anemia in β-thalassemia/HbE patients. Int J Hematol 104(3):338–343
Galanello R, Origa R (2010) Beta-thalassemia. Orphanet J Rare Dis 5(1):11
Ganz T (2011) Hepcidin and iron regulation, 10 years later. Blood 117(17):4425–4433
Yang Y, Li Y, Chen X, Cheng X, Liao Y, Yu X (2016) Exosomal transfer of miR-30a between cardiomyocytes regulates autophagy after hypoxia. J Mol Med 94(6):711–724
Acknowledgements
We would like to thank Tarbiat Modares University for its assistance and technical supports.
Author information
Authors and Affiliations
Contributions
MS, AA conceived and designed the experiments. MG, MA performed the experiments. AAZ contributed to sample preparation. MG, MA, MN, analyzed data and wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gholampour, M.A., Asadi, M., Naderi, M. et al. miR-30a regulates γ-globin expression in erythoid precursors of intermedia thalassemia through targeting BCL11A. Mol Biol Rep 47, 3909–3918 (2020). https://doi.org/10.1007/s11033-020-05483-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-020-05483-7