
Abstract
In higher plants, flower development is a result of crosstalk between many factors like photoperiod, vernalization, hormone concentration, epigenetic modification etc. and is also regulated by non-coding RNAs (ncRNAs). In the present study, we are reporting the involvement of long non-coding RNAs (lncRNAs) and miRNAs during the process of flower development in Cajanus scarabaeoides, an important wild relative of pigeonpea. The transcriptome of floral and leaf tissues revealed a total of 1672 lncRNAs and 57 miRNAs being expressed during flower development. Prediction analysis of identified lncRNAs showed that 1593 lncRNAs were targeting 3420 mRNAs and among these, 98 were transcription factors (TFs) belonging to 48 groups. All the identified 57 miRNAs were novel, suggesting their genera specificity. Prediction of the secondary structure of lncRNAs and miRNAs followed by interaction analysis revealed that 199 lncRNAs could interact with 47 miRNAs where miRNAs were acting in the root of interaction. Gene Ontology of the ncRNAs and their targets showed the potential role of lncRNAs and miRNAs in the flower development of C. scarabaeoides. Among the identified interactions, 17 lncRNAs were endogenous target mimics (eTMs) for miRNAs that target flowering-related transcription factors. Expression analysis of identified transcripts revealed that higher expression of Csa-lncRNA_1231 in the bud sequesters Csa-miRNA-156b by indirectly mimicking the miRNA and leading to increased expression of flower-specific SQUAMOSA promoter-binding protein-like (SPL-12) TF indicating their potential role in flower development. The present study will help in understanding the molecular regulatory mechanism governing the induction of flowering in C. scarabaeoides.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cajanus scarabaeoides (2n = 22) belongs to family Fabaceae and is a wild relative of pigeonpea (Cajanus cajan L.). It belongs to the secondary gene pool and is cross-compatible with the cultivated pigeonpea [1]. C. scarabaeoides possesses useful traits such as higher seed protein content, early flowering, tolerant to various biotic and abiotic stresses and used in the development of cytoplasmic male-sterile (CMS) systems [2]. Cultivated pigeonpea has a narrow genetic base and among them, only 3–4% is exploited for crop improvement [3]. On the other hand, CMS lines originated from C. scarabaeoides (A2 cytoplasm) has been successfully demonstrated potential for developing commercial hybrids [4]. Hence, C. scarabaeoides can serve as a potential donor for harnessing all desirable traits for the long-term sustainability of the pigeonpea crop.
Flower development is one of the essential events in the plant's life cycle and is directly related to seed set and yield [5]. It is a complex trait involving many genes and signaling pathways for the proper transition of the shoot apical meristem to flowering bud. The molecular mechanism behind flowering in plants has been explored in many crops including Arabidopsis [6] revealing the intricate interplay of genes. It is now known that only 2–3% of the genome codes for the protein-coding transcript which leads to functional proteins, while the rest of the transcripts are non-coding in nature, some of which gives rise to non-coding RNAs (ncRNAs). These ncRNAs play a major role in gene regulation [7]. Based on their length, they are classified into short non-coding RNAs (≤ 200 nucleotides) and long non-coding RNAs (≥ 200 nucleotides). Small nuclear RNAs (snRNAs), microRNAs (miRNAs), small interfering RNAs (siRNAs), small nucleolarRNAs (snoRNAs), transfer RNAs (t-RNAs), ribosomal RNAs (rRNAs), piwi-interacting RNAs (pi-RNAs) and trans-acting small interfering RNAs (tasi-RNAs) are the members of short non-coding RNAs family. On the other hand, long non-coding RNAs (lncRNAs) have 5′ cap and 3′poly A tail similar to coding mRNAs and are reported to be very large, running into hundreds of kb [8].
Based on the genomic position lncRNAs are mainly classified into four groups, long intergenic non-coding RNA (lincRNA), long intronic non-coding RNA, sense lncRNAs and antisense lncRNAs [9].These are not conserved among species and sometimes display a stage and tissue-specific expression pattern [9]. Long non-coding RNAs (lncRNAs) are one of the most diverse regulatory elements in the biological system, regulating at different levels during protein-coding gene expression. These lncRNAs can act as cis-acting elements (regulating a gene in its proximity) or trans-acting elements (regulating a distant gene). In most of the cases, it is trans-acting, inhibiting the transcription factor activity or preventing the binding of RNA polymerase II to the promoter (HOTAIR lncRNA silencing HOTAIR gene) [10]. Another lncRNA, Xist acted as a cis-acting element that was responsible for whole chromosome inactivation by epigenetic modifications [11].
Earlier, few studies have characterized the role of lncRNAs in plants and reported that COLDAIR (cold-assisted intronic non-coding RNA) and COOLAIR (cold-induced long antisense intragenic RNA) in Arabidopsis are involved in chromatin modification and silencing of the flowering locus C which leads to flower induction during the vernalization, showing their importance in plant growth and development [12, 13]. The lncRNAs are reported to play many important biological roles in the development and response to various environmental stresses [14]. Recently, numerous studies focusing on the role of lncRNAs in plants and their importance in various developmental processes and their response to various biotic and abiotic stresses have been studied in crops like Chinese cabbage, trifoliate orange, tobacco, tomato, rice, etc.[15,16,17,18,19].
LncRNA is known to act as endogenous target mimics (eTMs) of miRNAs as they contain competing miRNA-binding sites [20]. In this mechanism, lncRNAs mimics as the miRNA target and binds to it finally sequestering the miRNA and resulting in inhibition of the miRNA activity. The first lncRNA acting as eTM of miRNA was IPS1 in Arabidopsis thaliana [14]. Several studies on identification of lncRNAs acting as eTMs have been reported in crops like Arabidopsis, rice, soybean, tomato, cluster bean, black tea etc. [20,21,22,23,24].
MicroRNAs (miRNAs) are the members of small non-coding RNAs whose length varies from 20–24 nucleotides and regulate gene expression at the post-transcriptional level. These are transcribed into primary miRNAs (pri-miRNAs) by RNA polymerase II. The pri-miRNAs contain a stem-loop structure which is further processed by DICER-LIKE1 (DCL1) RNaseIII enzymes into a miRMA/miRNA* duplex. These miRNA/miRNA* duplexis exported to the cytoplasm with the help of exportin, where it associates with Argonaute (AGO) proteins and forms RNA-induced silencing complex (RISC) [25]. During the time of RISC loading, only one strand of the miRNA duplex is selected while the other strand is digested and removed by the exosome. This complex is guided by mature miRNA to search for complementary mRNA targets which leads to transcriptional and post-transcriptional gene silencing [26]. Various studies carried out on plants report that miRNAs are actively involved in most of the biological processes like growth and development, homeostasis response to abiotic and biotic stresses [27]. Identification and characterization of miRNAs in many crops like cotton, P. vulgaris, C. cajan, switch grass and potato have been reported [28,29,30,31,32]. Various studies on miRNAs have reported their role in vegetative to the reproductive phase transition, floral induction and flower development. In Arabidopsis thaliana it was reported that miR156 and miR172 were involved in regulating the timing of juvenile to adult transition; miR164 is involved in axillary bud formation and miR166 regulates shoot apical meristem and floral bud initiation [33,34,35].
However, to date lncRNAs and miRNAs have not been reported in C. scarabaeoides. Keeping this in view, the present study was undertaken to identify and characterize potential lncRNAs and miRNAs involved in the regulation of flower development in C. scarabaeoides. Furthermore, these findings will help to understand the regulatory role of lncRNAs and miRNAs during flowering in C. scarabaeoides.
Materials and methods
Genome-wide identification of lncRNAs
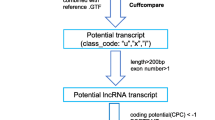
RNA sequence datasets from the leaf and bud tissues of C. scarabaeoides (SRX2661106, SRX2661107) were used for the present analysis [36]. The data were processed through Trimmomatic version 0.36 with default parameters [37]. Clean reads of each dataset were mapped on the reference pigeonpea genome (https://www.ncbi.nlm.nih.gov/assembly/GCF_000340665.1) separately with the help of Bowtie and Tophat 2.0 [38, 39]. All the mapped reads were assembled via the Cufflinks 2.0 [39] program separately for each dataset. All the predicted transcripts were merged to produce a consensus assembly using Cuffmerge software. Cuffdiff analysis was performed to calculate the transcript abundance and differential gene expression (log2 fold change ≥ 2 and ≤ -2, p-value > 0.05 and q-value > 0.01) in different tissues. Differential expression pattern of lncRNAs was represented using the MeV.4.8.1 software. Cuffcompare was also performed to obtain the class code of these transcripts. All transcripts with strand information were chosen for the downstream analysis whereas transcripts without strand information were discarded. The transcripts having FPKM value ≤ 0.5 were discarded while remaining sequences having length ≥ 200 bp and containing minimum one exon were selected for further analysis. All the selected transcripts were further processed through CPC (https://cpc.cbi.pku.edu.cn/) [40] and CNCI (https://www.bioinfo.org/software/cnci/) [41] programs to calculate their coding potential and the transcripts having coding potential > 0.5 were selected. The selected transcripts were further processed to identify whether they contain potential ORFs (open reading frame) via ORF Finder (https://www.ncbi.nlm.nih.gov/gorf/orfig.cgi) and Transdecoder (https://github.com/TransDecoder/TransDecoder.wiki.git). The transcripts having the potential to code for more than 100 amino acids were eliminated from our analysis. BLASTX (E-value cut-off of 1e-10, coverage ≥ 80% and identity ≥ 90%) against NCBI non-redundant (NR) protein database, KEGG database, COGs database, Swiss-Prot protein database and P-fam database were performed to rule out the transcripts having considerable homology to protein-coding gene. Finally, after the series of filtration, the remaining transcripts were expected to be lncRNAs. The lncRNAs prediction pipeline is given in (Supplementary Fig. 1).
Genomic conservation of C. scarabaeoides lncRNAs
To investigate the lncRNA conservation pattern, all lncRNA sequences were aligned against the genome sequences of T. aestivum, A. thaliana, Z. mays, G. max, C. arietinum, G. soja, V. unguiculata and V. radiata genomes with E-value < 1e-10. The similarity (≥ 20%) was set as the threshold for the identification of genome conservation.
Prediction of lncRNA targeted mRNAs
Both cis-acting and trans-acting targets were identified for lncRNAs. Gene present in 10 kb window of lncRNAs flanking region were considered as potential cis-target genes [42]. For trans-acting targets, the C. cajan mRNA database was used and the complementary mRNA sequence was predicted as a trans-acting target. We performed a BLAST search against the selected mRNAs sequences that were complementary to the lncRNA, setting E-value ≤ 1e-5 and identity ≥ 95%. The RNAplex software was used to calculate the complementary energy between lncRNAs and their targets with a cut-off value dNG -60 [43].
Genome-wide identification of miRNAs
To identify potential miRNAs, flowering stage leaf and bud transcriptome data of C. scarabaeoides was used. All known Viridiplantae miRNAs and pre-miRNAs were downloaded from miRBase22 (https://mirbase.org/). The pooled transcripts were BLASTN searched against the miRNAs and pre-miRNAs with an E-value cut-off ≤ 1e-3 and maximum of 3 nt mismatches. Both upstream (100 bp) and downstream (200 bp) regions along with the matched sequence were pooled out for further analysis. These sequences were then BLASTX search against C. cajan protein database with the similarity cut-off ≥ 80%; all the sequences retained in this cut-off were treated as potential protein-coding and were removed from the analysis. CPC and CNCI analysis were repeated with the remaining sequences to remove the potential-coding sequences. Leftover sequences were further filtered following the criteria described previously [29] to get predicted precursor pre-miRs sequences. The predicted sequences were selected and mature miRNAs were detected using mature-Bayes software. The pipeline followed for miRNAs prediction is represented in Supplementary Fig. 2.
Prediction of miRNA targeted mRNA/CDS
PsRNATarget (https://plantgrn.noble.org/psRNATarget/) was used to identify the prospective mRNAs/CDS as targets of miRNA [44]. To identify the probable mRNAs targeted by miRNAs, C. cajan CDS/mRNA sequences were used.
Prediction of lncRNA acting as miRNA target
The lncRNAs which can act as a probable target of miRNAs were identified by psRNA Target (https://plantgrn.noble.org/psRNATarget/) [44], with expectation value ≤ 3.5. The interaction network of lncRNAs and their prospective target genes were modeled with Cytoscape 3.2 [45].
Prediction of candidate lncRNAs for endogenous target mimics (eTMs)
The identification of lncRNAs acting as target mimics for miRNAs was performed by the method described previously [20]. The psRobot software (https://omicslab.genetics.ac.cn/psRobot/) was used to identify the putative eTMs. The secondary structures of lncRNAs and miRNAs were predicted with the Vienna RNA package RNA fold web (https://rna.tbi.univie.ac.at/).
Conservation analysis of lncRNAs
Conservation analysis of lncRNAs was performed by the BLASTN search with an E-value cut-off ≤ 1e-10 against the known lncRNAs from the CANTATA database (https://cantata.amu.edu.pl/) and NONCODE database (https://www.noncode.org/) of ncRNAs.
Gene ontology (GO) enrichment analysis
The possible functions of the identified targets of lncRNAs and targets of predicted miRNAs were determined using Gene Ontology (GO) program by setting significant enrichment (P ≤ 0.05) in the Blast2Go tool (https://www.blast2go.com/).
RNA isolation, quantification and cDNA synthesis
Spectrum plant total RNA kit (SIGMA) was used for total RNA isolation from the leaf and bud tissues of C. scarabaeoides followed by DNase (Ambion) treatment to remove DNA contamination. The quality and integrity of isolated RNA were tested on 1.2% denaturing agarose gel. RNA quantification was done using a spectrophotometer (Thermo Scientific). A good quality mRNA (~ 200 ng) was used for the first-strand cDNA synthesis using the Fermentas cDNA synthesis kit following the manufacturer’s protocol.
Validation of lncRNAs and their target genes using quantitative real-time PCR (qRT-PCR)
Expression analysis of prospective pigeon pea lncRNAs and their targets was performed using the leaf and bud tissues. Three technical replicates were taken for each sample in the qRT-PCR. The reaction was performed with a 1:10 ratio diluted cDNA to confirm the real-time amplification. All the primer sequence information is provided in Supplementary Table 1). The reaction mixture for qPCR analysis consisted of 12 µl Brilliant III Ultra-Fast SYBR Green QPCR Master Mix (ROX added) (Agilent Technologies, USA), 2 µl of diluted cDNA, 0.5 µl of each primer, and DEPC water. The reaction was carried out in a Light Cycler II qPCR system (Roche) and the reaction conditions were, 94 °C for 3 min, followed by 40 cycles of 94 °C for 30 s, 60 °C for 15 s and 72 °C for 20 s. This cycle was followed by a melting curve study ranging from 56 to 95 °C, with temperature increasing steps of 0.5 °C every 10 s. For each gene, α-tubulin was used as an internal control and the ‘comparative Ct method’ was used to calculate the gene expression [46].
miRNA isolation, cDNA synthesis and quantitative real-time PCR
miRNA was isolated from the leaf and bud tissues using RNASure Fusion miRNA Minikit (Genetix) according to the manufacturer’s protocol. The isolated small RNA was quantified using the Nanodrop spectrophotometer (Thermo Scientific). The cDNA synthesis was carried out with 1 µg of small RNA using the MirX miRNA First-Strand Synthesis kit (Clontech). The qRT-PCR analysis was performed using SYBR Advantage Premix (with ROX dye) in the Light Cycler II qPCR system (Roche) by protocol adopted by [47]. The relative expression pattern of each sample was analyzed by the ‘comparative Ct method’ [46]. Three technical replicates were used for each sample and U6 snRNA was used as an internal control.
Results and Discussion
Genome-wide identification of lncRNAs
C. scarabaeoides bud and leaf transcriptome data generated earlier by our lab (available as SRX2661106, SRX2661107) (https://www.ncbi.nlm.nih.gov/) were used for identification of lncRNAs and miRNAs. Total of 15,266,694 raw reads were processed using the Trimmomatic software. After processing and trimming, 14,423,287 (94.48%) high quality, clean reads were obtained. All the high-quality reads were mapped on to the C. cajan reference genome using TopHat2 and Bowtie. A total of 7,419,346 (51.44%) reads were mapped on C. cajan reference genome and were further used to estimate the abundance of the transcripts in terms of FPKM value using Cufflinks software. Cuffdiff analysis revealed that a total of 21,361 transcripts were expressed differentially in the bud and leaf tissues during flower development. The transcripts having length ≥ 200 bases, FPKM ≥ 0.5 and containing atleast one exon were filtered. Coding potential analysis of the remaining sequences was done via CPC and CNCI program resulting in 2246 sequences with CPC score ≤ 0. Further screening of the filtered sequences using Transdecoder and HMMER was done to remove the transcripts containing potential ORFs (sequence length more than 100 amino acid) and protein domain, followed by BLASTX analysis against Swissprot database resulting in the removal of 489 transcripts. Remaining 1757 transcript sequences were screened to identify sequence similarity with other groups of non-coding RNAs like tRNA, snRNA, snoRNA, r-RNA, etc. via BLASTN analysis. Finally, 1672 potential lncRNAs were identified and were carried forward for further analysis.
Characteristic features of the identified lncRNAs
The length of lncRNAs discovered in our study was found to be in the range of 200–10,000 bp having an average length of 2395 bp. Up to 31.57%, lncRNAs from our analysis were in the range of 200–1000 bp. Among the analyzed lncRNAs, 19.79% and 17.52% were found to be mono-exonic and di-exonic, respectively. The range of the exon number was found between 1 and 36. Surprisingly, only one lncRNAs was having 36 exons and rest fall between 1 and 20 exons category. Predicted lncRNAs were found to be AU rich when compared to the mRNAs. The mean GC content was 37.99%, lower than that of coding sequences (42.33%) of C. cajan. The result is in accordance with the lncRNAs discovered in other crops like Arabidopsis, maize and rice [15, 48, 49]. The chromosome-wise distribution illustrates that the highest numbers of lncRNAs (264) were present on chromosome 11 while 280 lncRNAs remained unmapped on the chromosomes but still belonged to unassigned scaffolds. The detailed physical information like length, GC/AU percentage, exon number, and genomic location of the predicted lncRNAs is provided in Fig. 1 and Supplementary Table 2.

Physical properties of predicted lncRNAs in Cajanus scarabaeoides. a Chromosome wise distribution of lncRNAs, b Length distribution of lncRNAs, c Exon number distribution of lncRNAs, d Comparative distribution GC% and AU% in lncRNAs and mRNAs
Conservation analysis of lncRNAs with other species
Conservation analysis of the lncRNAs confirmed that all the lncRNAs except two (Csa-lncRNA_425, Csa-lncRNA_1189) identified in this study were novel and specific to C. scarabaeoides. For genome conservation, lncRNAs showing a ≥ 20% sequence similarity with other genomes were considered conserved. The results revealed that the 91.74% and 91.38% lncRNAs were conserved with G. soja and G. max, respectively. Followed by 81.86% 80.86%, 57.83% lncRNAs were conserved with V. unguiculata, V. radiate and C. arietinum. Only a few lncRNAs showed conservation with A. thaliana (5.26%), T.aestivum (4.72%) and Z. mays (3.76%). It was observed that the lncRNAs identified in this study were highly conserved amongst the legumes and were less conserved with distant genera. The finding was supported by [50], stating that legumes lncRNAs showed low conservation with the distant genera.
Differential expression profiling of lncRNAs between leaf and bud tissues
A total of 1672 lncRNAs were found to be expressed in the leaf and bud tissues of C. scarabaeoides during flower development. Only 368 lncRNAs were differentially expressed between the leaf and bud tissues. Among 368 differentially expressed lncRNAs, 143 were down-regulated and 225 were up-regulated in the buds as compared to leaves. The lncRNAs expression in each tissue varied from log2 fold 8.48 to − 6.99 (Fig. 2a, b and Supplementary Table 3). Among the differentially expressed lncRNAs, only 1 lncRNA was bud specific while 10 were leaf specific. The differentially expressed lncRNAs have a high tissue-specificity index, which supports their tissue-specific expression pattern.

a Differentially expressed lncRNAs in Cajanus scarabaeoides, X-axis represent lncRNAs and Y-axis represent log2 fold change expression in the bud in comparison to leaf, b number of up and down-regulated lncRNAs in the bud compared to leaf, c number of transcription factors (TFs) targeted by lncRNAs
Identification of lncRNAs targeted mRNA
LncRNAs control the gene expression by positively regulating the target gene by sequestering gene-specific miRNA, or accommodating other regulatory elements in 5′UTRs or the genes and regulate gene expression both positively and negatively. The super-secondary structure of the lncRNAs provides binding sites for miRNA, mRNA, TFs and other regulatory elements. A total of 1593 lncRNAs have potential binding sites for 3420 mRNAs and among these 98 were TFs belonging to 48 TFs groups (Fig. 2c and Supplementary Table 4).
GO annotation of lncRNAs targeted mRNAs
Gene ontology (GO) annotation of the lncRNAs and lncRNA targeted mRNAs were performed and the ones with P-value ≤ 0.05 were considered significant GO terms. The results were further classified into three categories: biological processes, molecular functions and cellular components (Supplementary Table 5).Under the Biological processes, the majority of the lncRNAs (47.47%) were involved in the metabolic and cellular process. Around 29.15% of the lncRNAs were involved in the single-organism processes and biological regulations, and 18.96% of the lncRNAs were associated with localization, response to a stimulus, signaling and cellular component organization or biogenesis. The rest (4.39%) were involved in the developmental process, multicellular organismal process, detoxification, and reproductive process, positive and negative regulation of the biological process, multi-organism process, growth, and apoptosis.
In molecular functions, the majority of the lncRNAs performed functions in catalytic activity (44.21%) and binding (40.18%). Around 13.82% were involved in structural molecule activity, transporter activity, nucleic acid binding transcription factor activity, antioxidant activity, transcription factor activity, protein binding. Rest 1.78% were associated with electron carrier activity, signal transducer activity, molecular transducer activity, nutrient reservoir activity and protein tag. The lncRNAs were localized in different cellular components, the majority were involved in cell and cell part (40.31%), membrane and membrane parts (28.39%). Around 30.47% were associated with organelle, macromolecular complex, organelle part, and membrane-enclosed lumen. The rest were localized in the extracellular region, supra-molecular complex, cell junction, symplast, extracellular region part, virion, virion part and nucleoid (Supplementary Table 5).
Distribution of the mRNAs based on biological processes revealed that the majority of lncRNAs were involved in metabolic and cellular processes (47.47%) followed by biological regulation (29.15%) which are the basic activities and are required for survival of the plants. The third and most important class i.e. biological regulation is represented by around 19% of total lncRNAs which directly deal with flower development by sensing different stimuli, regulates signaling and is involved in the biogenesis of different organs. Under molecular functions, around 14% of lncRNAs targeted mRNAs were found to be involved in structural molecule activity, transporter activity, nucleic acid binding transcription factor activity, antioxidant activity and transcription factor activity which are involved in the development of new structures like flowers and buds (Supplementary Fig. 3, Supplementary Table 5). Through GO analysis we can conclude that 10 to 20% of total lncRNAs may have a putative role in flower development processes.
Genome-wide identification of miRNAs in C. scarabaeoides
A total of 57 miRNAs were identified from the leaf and bud transcriptome data of C. scarabaeoides. All the predicted miRNAs in this study were found to be novel with a length of 22 nucleotides. The physical properties of these predicted miRNAs are provided in Supplementary Table 6. The identified miRNAs represent 35 different miRNA families with 12 diverse SSR signatures. The predominant miRNA families were miR156, miR166, miR168, miR390, miR408, miR171, and miR495 (Supplementary Table 7).
Identification of mRNA targeted bymiRNAs
miRNAs control the gene expression at both transcriptional and post-transcriptional level and further many non-coding RNAs are also involved in miRNA mediated transcriptional gene silencing. Keeping this in the background we searched for the targets of the identified miRNAs using the psRNA Target server. To identify mRNA as a target, the mRNA/CDS sequence of C. cajan was used as a subject with 3.5 mismatches threshold value. It was found that 57 miRNAs targeted 1874 mRNAs including 69 transcription factors (TFs) belonging to 22 families (Supplementary Table 8 and Supplementary Fig. 4). The identified TFs were MYB, Ethylene responsive factors, GTE, bHLH, MADS-box, TCP, NAC, WRKY, LHW, PIF etc. which are majorly involved in flowering. Further, we identified the interaction patterns of the mRNAs, transcription factors and miRNAs through the cytoscape (Fig. 3a).

miRNA targeted mRNAs including transcription factors (TFs), center of the interaction represents miRNA and in periphery the hexagonal red box represent the mRNAs and yellow hexagonal blocks represents the TFs. a miR168b-GATA TF interaction, b miR821MADS box TF interaction, c miR4383a-GTE8 TF and miR4383a-bHLH TF interaction, d miR481a-MYB TF interaction. (Color figure online)
The interaction analysis revealed that Cc-miR168b was interacting with many mRNAs like XM_020379752.1 (RDM-16 like), XM_020355987.1 (uncharacterized), XM_020373785.1 (pentatricopeptide repeat-containing protein), XM_020375660.1 (GATA transcription factor 26-like) and XM_020361814.1 (GATA transcription factor 26-like). In Arabidopsis, two paralogs of GATA transcription factors GNC and GNC-LIKE (GNL)/CGA1 which acts downstream to Auxin responsive factor (ARF2) controlling greening, flowering time and senescence were reported [51].This suggests that Cc-miR168b may have a putative role in flower development pathways mediated through hormones in C. scarabaeoides.
Another miRNA i.e. Cc-miR821 was found to be interacting with many mRNAs among them XM_020384773.1 (MADS-box transcription factor 23 like), XM_020384775.1 (MADS-box transcription factor 23 like), XM_020384776.1 (MADS-box transcription factor 23 like), XR_002241500.1 (MADS-box transcription factor 23 like) and XR_002241501.1(MADS-box transcription factor 23 like) belongs to MADS-box TF family (Fig. 3b) which have significant role in controlling development of flower, embryo, fruits, seed and root [52]. The results revealed that Cc-miR4383a interacted with bHLH TFs as well as a GTE8 transcription factor (Fig. 3c). It was seen that miR482 interacted with MYB transcription factors along with other mRNAs (Fig. 3d). MYB family of transcription factors plays important roles in the regulation of plant development, hormone signaling, defense response and secondary metabolism. The role of MYB305 and MYB340 in regulating the expression of flavonoid biosynthetic genes and flower development in antirrhinum flowers was reported by [53]. Similarly, Gh-MYB8 TF was found to be interacting with bHLH TF GMYC1 and helping in the activation of a late anthocyanin biosynthetic gene promoter PGDFR2 [54]. Thus, from our results, we can conclude that these TFs play a significant role in flower development and are regulated by respective miRNAs.
GO annotation of miRNAs targeted mRNAs
GO annotation results were classified into biological processes, molecular functions and cellular components categories (Supplementary Fig. 5). Under the Biological processes, the majority of the targets (55.53%) were involved in the metabolic and cellular processes. Around 42.24% of the targets were involved in biological regulation, regulation of the biological process, single-organism process, response to a stimulus, localization, signaling and cellular component organization or biogenesis. In molecular functions, the majority of the targets performed functions in binding (46.77%) and catalytic activity (40.33%). Around 12.88% were involved in transporter activity, nucleic acid binding transcription factor activity, signal transducer activity, molecular transducer activity, molecular function regulator, protein binding and transcription factor activity. The target mRNAs were found to be localized in different cellular components, among them a large number of proteins localize in cell and cell part (64.79%) and organelle (21.16%). Distribution revealed that the majority of the miRNA targeted mRNAs were involved in common cellular functions. But among the biological functions (42.24%) and molecular functions (12.88%), the mRNAs were involved in pathways that may be putatively acting in flower development (Supplementary Table 9).
Involvement of lncRNAs and their potential target genes in flower regulation
lncRNAs are directly involved in gene expression acting in both cis and trans-manner, it recruits transcription factors, epigenetic modifiers and/or inhibitors, RNA polymerase II which up-regulate or down-regulate the expression of the target gene [55]. In the present study, genes for the predicted lncRNAs and their targets were identified and analyzed for their expression pattern through real-time PCR. The gene XM_020357923.1 codes for protein pollen-specific allergen and is targeted by Csa-lncRNA_328, and showed high expression in bud compared to leaf and expression pattern of its corresponding lncRNA showed a positive correlation. Similarly, the genes XM_020361200.1 (Esterase C25G4.2 like) targeted by Csa-lncRNA_592, XM_020366137.1 (a E3 ubiquitin-protein ligase) targeted by Csa-lncRNA_881 and XM_020371981.1 (a extracellular ribonuclease LE-like protein) targeted by Csa-lncRNA_1221 showed higher expression in bud as compared to the leaf and their corresponding lncRNAs also followed the similar pattern. XM_020363582.1 (TIFY6B like protein expressed in inflorescence meristem) targeted by Csa-lncRNA_836 and XM_020373686.1 (a gibberellin-regulated protein 1) targeted by Csa-lncRNA_1412, both the genes and their corresponding lncRNAs were highly expressed in the leaf and repressed in the bud tissue. The results revealed that lncRNAs and their target mRNAs have similar expression pattern in specific tissues, pointing towards their involvement in positive regulation of their target genes (Fig. 4).

Real-time PCR analysis of the predicted lncRNAs and their targets in Cajanus scarabaeoides, Y-axis represent relative expression (log2 fold) of lncRNAs and their mRNA targets and X-axis represent the tissues used for the real-time analysis
lncRNAs and miRNA interaction analysis
psRNATarget server was utilized for the prediction of lncRNAs and miRNAs interaction. A total of 199 lncRNAs were targeted by 47 miRNAs (mismatches allowed up to 3.5) (Supplementary Table 10). The secondary structure of lncRNAs and miRNAs were also predicted to identify interaction sites between them. The interaction pathway between 199 lncRNAs with 47 miRNAs was visualized by Cytoscape (Supplementary Fig. 6). It was observed that the miRNAs were always present in the root of the interaction pattern which revealed that miRNAs were the main regulatory elements in this network that seems to be associated with flowering in C. scarabaeoides. Similar kind of results were also reported in chickpea [56].
The Cytoscape interaction studies between miRNAs and lncRNAs revealed that the members of the miRNAs families like miR-169, miR-172, miR-530, miR-495, miR-156, miR396, miR-408, miR-166 and miR-319 were actively interacting with lncRNAs. The members of Csa-miR156 family interacted with Csa-lncRNA_440, Csa-lncRNA_515, Csa-lncRNA_607, Csa-lncRNA_651, Csa-lncRNA_1425 and Csa-lncRNA_1585. Similarly, the members of Csa-miR166 family interacted with Csa-lncRNA_534, Csa-lncRNA_892, Csa-lncRNA_1114 and Csa-lncRNA_1606. The members of Csa-miR172 interacted with Csa-lncRNA_664, Csa-lncRNA_784, Csa-lncRNA_970 and Csa-lncRNA_1317. These miRNAs are already reported to be associated with flower development, photoperiodism, stress response, auxin activated signaling pathways [56].
Prediction of endogenous target mimics (eTMs)
Endogenous target mimics (eTMs) are lncRNAs known for target mimicry with the sequestered transcripts and results in inhibition of the miRNA expression [57]. Endogenous target mimics (eTMs) for the predicted miRNAs were identified via psRobot software. The eTMs prediction results showed that 17 lncRNAs were binding to miRNAs with minimum fold energy (Fig. 5, Supplementary Sheet 1). Interestingly, all these 17 lncRNAs interacted with miRNAs whose targets are transcription factors mainly involved in flower development (Supplementary Table 11). Hence it can be hypothesized that these 17 lncRNAs indirectly regulate the function of these transcription factors by the mechanism of endogenous target mimic (eTMs) via miRNAs. For example, Csa-lncRNA_1231 regulates the expression of SPL family transcription factors via Csa-miR156. In the cell, more concentration of Csa-miR156 can suppress the expression of SPL regulatory genes which leads to late flowering phenotype in Arabidopsis [33]. Simultaneously, more expression of Csa-lncRNA_1231 in the cell having an alternate target/binding site for Csa-miR156 results in a higher concentration of SPL family transcription factor resulting in early flowering. Previously, eTMs were identified in the degradome data of maize with 34 lncRNAs having a binding site for 33 miRNAs involved in the regulation of various pathways. The study reported that the expression of lncRNAs disrupted the miRNA-mRNA regulation [58].

a Predicted base pairing interactions between endogenous target mimic (eTM) lncRNAs (green color) and miRNA (red color), b Real-time expression analysis of eTMs (Csa-lncRNA_1231 and Csa-miRNA156b) along with target transcription factors SPL-12 (Squamosa binding like protein) in the leaf and bud tissues. (Color figure online)
It was also observed that in some of the cases two or more than two Csa-lncRNAs were targeted by a single Csa-miRNA. Csa-lncRNA_1231 and Csa-lncRNA_1543 were endogenously targeted by Csa-miR156b and Csa-miR156c respectively. Similarly, Csa-lncRNA_122 and Csa-lncRNA_866 were targeted by Csa-miR495b; Csa-lncRNA_1416 and Csa-lncRNA_1499 targeted by Csa-miR169; Csa-lncRNA_1375, Csa-lncRNA_711 and Csa-lncRNA_1445 targeted by Csa-miR530a; Csa-lncRNA_704, Csa-lncRNA_1662 and Csa-lncRNA_721 targeted by Csa-miR818. Similar kind of interaction pattern of the eTMs has been reported in Cyamopsis tetragonoloba [23].
Validation of lncRNAs, miRNAs and e-TMs targets through qRT-PCR
The results obtained through a computational analysis were validated through quantitative real-time PCR using leaf and bud tissues of C. scarabaeoides. For validation, Csa-lncRNA_1231, its interacting Csa-miR156b and target transcription factor SPL12 were used. The results revealed that in leaf, expression of Csa-lncRNA_1231 and SPL 12 was low, and Csa-miR156b was highly expressed. In flower bud, expression of Csa-lncRNA_1231 and SPL 12 were higher and Csa-miR156b showed lower expression. In leaf, higher expression of Csa-miR156b, degraded SPL12 specific transcript which decreases its concentration in the leaf. But in the flower bud, presence of Csa-lncRNA_1231 sequester maximum amount of Csa-miR156b, hence Csa-miR156b is unavailable to degrade transcript of SPL12 and hence the concentration of SPL 12 increases which leads to initiation of flowering and flower development (Fig. 5). Previous reports on lncRNAs have validated only the lncRNAs and miRNAs but here in this study, we tried to validate the effect of eTMs on the targeted genes. The results revealed that in the presence of corresponding Csa-lncRNA, the miRNA level in the tissues was reduced which results in the expression of miRNA targeted genes.
Conclusion
Cajanus scarabaeoides is an important wild relative of pigeonpea acting as a good source for traits like early flowering, male sterility, and resistance to biotic and abiotic stresses. Non-coding RNAs (miRNAs and lncRNAs) play an important role as a regulatory element in gene expression. In this study, a total of 1,672 lncRNAs and 57 miRNAs were identified utilizing transcriptome data of the bud and leaf tissues. Majority of the lncRNA were differentially expressed among the tissues but only one was bud specific and 10 were leaf specific. Most of these ncRNAs affect gene regulation by interacting with transcription factors, which is the key to their involvement in various developmental processes. The lncRNAs acting as both cis and trans-acting elements were found to have an important role in flower development in C. scarabaeoides. Interactome analysis reveals that miRNA is the key player and stands in the roots of all interactions between miRNA, mRNA and lncRNA. It was observed that 17 Csa-lncRNA were interfering with Csa-miRNA, therefore acting as important eTMs. Although it has been reported that lncRNAs can both up-regulate and down-regulate the gene expression but we found only those lncRNAs which can increase the gene expression in a tissue-specific manner e.g. Csa-lncRNA_328, Csa-lncRNA_592, Csa-lncRNA_881 and Csa-lncRNA_1221. These lncRNAs were found to be mimicking the targets of miRNA and our results were in accordance with this. The Csa-lncRNA1231 indirectly regulates the expression flower development-specific transcription factor SPL12 by sequestering SPL12 specific Csa-miR156b. To our knowledge, this is a detailed report on identification and characterization of lncRNAs and miRNAs involved in flowering in C. scarabaeoides. Therefore, the present study will provide a basis to understand the regulatory network of non-coding RNAs in flower development in C. scarabaeoides.
Abbreviations
- AGO:
-
Argonaute
- ARF:
-
Auxin responsive factor
- BLAST:
-
Basic local alignment search tool
- bp:
-
Base pairs
- cDNA:
-
Complementary DNA
- CMS system:
-
Cytoplasmic male-sterile system
- COG:
-
Clusters of orthologous groups
- eTMs:
-
Endogenous target mimics
- GO:
-
Gene ontology
- kb:
-
Kilo base
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- lincRNAs:
-
Long intergenic non-coding RNAs
- lncRNAs:
-
Long non-coding RNAs
- min:
-
Minutes
- miRNAs:
-
MicroRNAs
- mRNAs:
-
Messenger RNAs
- NATs:
-
Natural antisense transcripts
- ncRNAs:
-
Non-coding RNAs
- Nr database:
-
Non-redundant protein database
- nt:
-
Nucleotides
- ORF’s:
-
Open reading frames
- PCR:
-
Polymerase chain reaction
- pi-RNAs:
-
Piwi-interacting RNAs
- pri-miRNAs:
-
Primary miRNAs
- qRT-PCR:
-
Quantitative real time PCR
- RISC:
-
RNA-induced silencing complex
- r-RNAs:
-
Ribosomal RNAs
- sec:
-
Seconds
- siRNAs:
-
Small interfering RNAs
- snoRNAs:
-
Small nucleolar RNAs
- snRNAs:
-
Small Nuclear RNAs
- SPL:
-
SQUAMOSA promoter-binding protein like
- tasi-RNAs:
-
Trans-acting small interfering RNAs
- TF’s:
-
Transcription factors
- t-RNAs:
-
Transfer RNAs
- UTRs:
-
Untranslated regions
References
Saxena KB (2008) Genetic improvement of pigeon pea-a review. Trop Plant Biol 1:159–178. https://doi.org/10.1007/s12042-008-9014-1
Khoury CK, Castañeda-Alvarez NP, Achicanoy HA et al (2015) Crop wild relatives of pigeon pea (Cajanus cajan (L.) Millsp.): distributions, ex-situ conservation status, and potential genetic resources for abiotic stress tolerance. Biol Conserv 184:259–270. https://doi.org/10.1016/j.biocon.2015.01.032
Kassa MT, Penmetsa RV, Carrasquilla-Garcia N et al (2012) Genetic patterns of domestication in pigeonpea (Cajanus cajan (L.) Millsp.) and wild Cajanus relatives. PLoS ONE 7:e39563. https://doi.org/10.1371/journal.pone.0039563
Choudhary AK, Singh IP (2015) A study on comparative fertility restoration in A2 and A4 cytoplasms and its implication in breeding hybrid pigeonpea [Cajanus cajan (L.) Millspaugh]. AJPS 6:385–391. https://doi.org/10.4236/ajps.2015.62044
Yoo SY, Kardailsky I, Lee JS et al (2004) Acceleration of flowering by overexpression of MFT (Mother of FT and TFL1). Mol Cells 17:95–101
Hong Y, Jackson S (2015) Floral induction and flower formation-the role and potential applications of miRNAs. Plant Biotechnol J 13:282–292. https://doi.org/10.1111/pbi.12340
Bai Y, Dai X, Harrison AP, Chen M (2015) RNA regulatory networks in animals and plants: A long noncoding RNA perspective. Brief Funct Genomics 14:91–101. https://doi.org/10.1093/bfgp/elu017
Guttman M, Amit I, Garber M et al (2009) Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458:223–227. https://doi.org/10.1038/nature07672
Ma L, Bajic VB, Zhang Z (2013) On the classification of long non-coding RNAs. RNA Biol 10:924–933. https://doi.org/10.4161/rna.24604
Rinn JL, Kertesz M, Wang JK et al (2007) Functional demarcation of active and silent chromatin domains in human HOX loci by non-coding RNAs. Cell 129:1311–1323. https://doi.org/10.1016/j.cell.2007.05.02
Maenner S, Blaud M, Fouillen L et al (2010) 2-D structure of the A region of Xist RNA and its implication for PRC2 association. PLoS Biol 8:e1000276. https://doi.org/10.1371/journal.pbio.1000276
Swiezewski S, Liu F, Magusin A, Dean C (2009) Cold-induced silencing by long antisense transcripts of an Arabidopsis Polycomb target. Nature 462:799–802. https://doi.org/10.1038/nature0861
Heo JB, Sung S (2011) Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA. Science 331:76–79. https://doi.org/10.1126/science.1197349
Franco-Zorrilla JM, Valli A, Todesco M et al (2007) Target mimicry provides a new mechanism for regulation of microRNA activity. Nat Genet 39:1033–1037. https://doi.org/10.1038/ng2079
Wang A, Hu J, Gao C et al (2019) Genome-wide analysis of long non-coding RNAs unveils the regulatory roles in the heat tolerance of Chinese cabbage (Brassica rapassp. chinensis). Sci Rep 9:5002. https://doi.org/10.1038/s41598-019-41428-2
Wang CY, Liu SR, Zhang XY et al (2017) Genome-wide screening and characterization of long non-coding RNAs involved in flowering development of trifoliate orange (Poncirustrifoliata L. Raf.). Sci Rep 7:43226. https://doi.org/10.1038/srep43226
Li X, Xing X, Xu S et al (2018) Genome-wide identification and functional prediction of tobacco lncRNAs responsive to root-knot nematode stress. PLoS ONE 13:e0204506. https://doi.org/10.1371/journal.pone.0204506
Zhou Y, Cho WK, Byun HS et al (2019) Genome-wide identification of long non-coding RNAs in tomato plants irradiated by neutrons followed by infection with Tomato yellow leaf curl virus. PeerJ 7:e6286. https://doi.org/10.7717/peerj.6286
Zhao J, Ajadi AA, Wang Y et al (2020) Genome-wide identification of lncRNAs during rice seed development. Genes 11:243. https://doi.org/10.3390/genes11030243
Wu HJ, Wang ZM, Wang M et al (2013) Wide spread long non-coding RNAs as endogenous target mimics for microRNAs in plants. Plant Physiol 161:1875–1884. https://doi.org/10.1104/pp.113.215962
Ye CY, Xu H, Shen E et al (2014) Genome-wide identification of non-coding RNAs interacted with microRNAs in soybean. Front Plant Sci 5:743. https://doi.org/10.3389/fpls.2014.00743
Wang J, Yu W, Yang Y et al (2015) Genome-wide analysis of tomato long non-coding RNAs and identification as endogenous target mimic for microRNA in response to TYLCV infection. Sci Rep 5:16946. https://doi.org/10.1038/srep16946
Sahu S, Rao AR, Pandey J et al (2018) Genome-wide identification and characterization of lncRNAs and miRNAs in cluster bean (Cyamopsis tetragonoloba). Gene 667:112–121. https://doi.org/10.1016/j.gene.2018.05.027
Varshney D, Rawal HC, Dubey H et al (2019) Tissue specific long non-coding RNAs are involved in aroma formation of black tea. Ind Crops Prod 133:79–89. https://doi.org/10.1016/j.indcrop.2019.03
Reinhart BJ, Weinstein EG, Rhoades MW et al (2002) MicroRNAs in plants. Genes Dev 16:1616–1626. https://doi.org/10.1101/gad.1004402
Filipowicz W, Bhattacharyya SN, Sonenberg N (2008) Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet 9:102–114. https://doi.org/10.1038/nrg2290
Navarro L, Dunoyer P, Jay F et al (2006) A plant miRNA contributes to antibacterial resistance by repressing auxin signaling. Science (New York, NY) 312:436–439. https://doi.org/10.1126/science.1126088
Zhang B, Wang Q, Wang K et al (2007) Identification of cotton microRNAs and their targets. Gene 397:26–37. https://doi.org/10.1016/j.gene.2007.03.020
Nithin C, Patwa N, Thomas A et al (2015) Computational prediction of miRNAs and their targets in Phaseolus vulgaris using simple sequence repeat signatures. BMC Plant Biol 15:1–16. https://doi.org/10.1186/s12870-015-0516-3
Nithin C, Thomas A, Basak J, Bahadur RP (2017) Genome-wide identification of miRNAs and lncRNAs in Cajanus cajan. BMC Genomics 18:1–14. https://doi.org/10.1186/s12864-017-4232-2
Matts J, Jagadeeswaran G, Roe BA, Sunkar R (2010) Identification of microRNAs and their targets in switch grass, a model biofuel plant species. J Plant Physiol 167:896–904. https://doi.org/10.1016/j.jplph.2010.02.001
Xie F, Frazier TP, Zhang B (2011) Identification, characterization and expression analysis of microRNAs and their targets in the potato (Solanumtuberosum). Gene 473:8–22. https://doi.org/10.1016/j.gene.2010.09.007
Wu G, Park MY, Conway SR et al (2009) The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell 138:750–759. https://doi.org/10.1016/j.cell.2009.06.031
Raman S, Greb T, Peaucelle A et al (2008) Interplay of miR164, cup-shaped cotyledon genes and lateral suppressor controls axillary meristem formation in Arabidopsis thaliana. Plant J 55:65–76. https://doi.org/10.1111/j.1365-313X.2008.03483
Jung JH, Park CM (2007) MIR166/165 genes exhibit dynamic expression patterns in regulating shoot apical meristem and floral development in Arabidopsis. Planta 225:1327–1338. https://doi.org/10.1007/s00425-006-0439-1
Nigam D, Saxena S, Ramakrishna G et al (2017) De novo assembly and characterization of Cajanus scarabaeoides (L.) Thouars Transcriptome by paired-end sequencing. Front Mol Biosci 4:48. https://doi.org/10.3389/fmolb.2017.00048
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Kim D, Pertea G, Trapnell C et al (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14:R36. https://doi.org/10.1186/gb-2013-14-4-r36
Trapnell C, Roberts A, Goff L et al (2012) Differential gene and transcript expression analysis of RNA-Seq experiments with top hat and cufflinks. Nat Protoc 7:562–578. https://doi.org/10.1038/nprot.2012.016
Kong L, Zhang Y, Ye ZQ et al (2007) CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res 35:345–349. https://doi.org/10.1093/nar/gkm391
Sun L, Luo H, Bu D et al (2013) Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res 41:e166. https://doi.org/10.1093/nar/gkt646
Jia H, Osak M, Bogu GK et al (2010) Genome-wide computational identification and manual annotation of human long noncoding RNA genes genome-wide computational identification and manual annotation of human long noncoding RNA genes. RNA 16:1478–1487. https://doi.org/10.1261/RNA.1951310
Tafer H, Hofacker IL (2008) RNAplex: A fast tool for RNA-RNA interaction search. Bioinformatics 24:2657–2663. https://doi.org/10.1093/bioinformatics/btn193
Dai X, Zhuang Z, Zhao PX (2018) PsRNATarget: a plant small RNA target analysis server (2017 release). Nucleic Acids Res 46:W49–W54. https://doi.org/10.1093/nar/gky316
Saito R, Smoot ME, Ono K, Ruscheinski J (2013) A travel guide to Cytoscape plugins. Nat Methods 9:1069–1076. https://doi.org/10.1038/nmeth.2212
Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative CTmethod. Nat Protoc 3:1101–1108. https://doi.org/10.1038/nprot.2008.73
Tyagi A, Nigam D, Mithra SVA et al (2018) Genome-wide discovery of tissue-specific miRNAs in cluster bean (Cyamopsis tetragonoloba) indicates their association with galactomannan biosynthesis. Plant Biotechnol J 16:1241–1257. https://doi.org/10.1111/pbi.12866
Di Rubbo S, Irani NG, Kim SY et al (2013) The clathrin adaptor complex AP-2 mediatesendocytosis of BRASSINOSTEROID INSENSITIVE1 in Arabidopsis. Plant Cell 25:2986–2997. https://doi.org/10.1105/tpc.113.114058
Zhang W, Han Z, Guo Q et al (2014) Identification of maize long non-coding RNAs responsive to drought stress. PLoS ONE 9:98958. https://doi.org/10.1371/journal.pone.0098958
Wen J, Parker BJ, Weiller GF (2007) In Silico identification and characterization of mRNA-like non-coding transcripts in Medicago truncatula. Silico Biol 7:485–505
Richter R, Bastakis E, Schwechheimer C (2013) Cross-repressive interactions between SOC1and the GATAs GNC and GNL/CGA1 in the control of greening cold tolerance and flowering time in Arabidopsis. Plant Physiol 162:1992–2004. https://doi.org/10.1104/pp.113.219238
Masiero S, Colombo L, Grini PE et al (2011) The emerging importance of Type I MADSBox transcription factors for plant reproduction. Plant Cell 23:865872. https://doi.org/10.1105/tpc.110.081737
Moyano E, Martinez-Garcia JF, Martin C (1996) Apparent redundancy in MYB gene function provides gearing for the control of flavonoid biosynthesis in Antirrhinum flowers. Plant Cell 8:1519–1532. https://doi.org/10.1105/tpc.8.9.1519
Laitinen RAE, Immanen J, Auvinen P et al (2005) Analysis of the floral transcriptome uncovers new regulators of organ determination and gene families related to flower organ differentiation in Gerbera hybrida (Asteraceae). Genome Res 15:475–486. https://doi.org/10.1101/gr.3043705
Wang KC, Chang HY (2011) Molecular mechanisms of long noncoding RNAs. Mol Cell 43:904–914. https://doi.org/10.1016/j.molcel.2011.08.018
Khemka N, Singh VK, Garg R, Jain M (2016) Genome-wide analysis of long intergenic non-coding RNAs in chickpea and their potential role in flower development. Sci Rep 6:33297. https://doi.org/10.1038/srep33297
Bohra P, Das A, Milner MJ et al (2018) Long non-coding rnas as endogenous target mimics and exploration of their role in low nutrient stress tolerance in plants. Genes (Basel) 9:459. https://doi.org/10.3390/genes9090459
Fan C, Hao Z, Yan J, Li G (2015) Genome-wide identification and functional analysis of lincRNAs acting as miRNA targets or decoys in maize. BMC Genomics 16:793. https://doi.org/10.1186/s12864-015-2024-0
Acknowledgements
The authors acknowledge the support received from the Director, ICAR-National Institute for Plant Biotechnology, NewDelhi.
Funding
We acknowledge the financial support received from ICAR-National Institute for Plant Biotechnology, New Delhi, India.
Author information
Authors and Affiliations
Contributions
Experiments were designed by KG and NKS; AD and SS conducted experiments; and all authors analyzed, wrote and edited the manuscript. All authors read and approve the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Authors declare no conflict of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Das, A., Saxena, S., Kumar, K. et al. Non-coding RNAs having strong positive interaction with mRNAs reveal their regulatory nature during flowering in a wild relative of pigeonpea (Cajanus scarabaeoides). Mol Biol Rep 47, 3305–3317 (2020). https://doi.org/10.1007/s11033-020-05400-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-020-05400-y