
Abstract
Sulfonamide derivatives, such as sulfadiazine (SD) are used as antibiotics and, very recently, anti-amyloid properties of these have been reported. We have evaluated binding of SD with bovine serum albumin (BSA) followed by unfolding of protein. Studies were accomplished at physiological conditions of temperature (37 °C) and pH (7.4), employing UV, fluorescence, circular dichroism (CD) and Fourier transform infra-red (FTIR) spectroscopies. In presence of drug, UV spectrum of BSA was altered from the spectrum of native BSA due to the interaction between albumin and drug. Excitation of protein at 295 nm showed that fluorescence quenching of BSA by SD is a result of the formation of SD–BSA complex. The data were analyzed using Stern–Volmer and Lineweaver–Burk methods. From both methods it was evaluated that the quenching involved in BSA–SD binding was static. BSA had only one binding site for SD. Synchronous fluorescence spectra have shown a red shift and advocated that hydrophobicity around both Trp and Tyr residues was decreased. CD results revealed that the conformation of macromolecule remain undisturbed at low concentrations (up to 20 μM of the SD) and there was small perturbation in the secondary structure from 20 to 50 μM of SD followed by a large change and consequent unfolding on further increase in the drug concentration. Both synchronous and CD measurements were consistent to each other. FTIR spectra revealed the shifting of amide I band which is also an indication of conformational change of the protein.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
For the structure-based drug design, a meticulous understanding of intermolecular interactions amongst proteins and their ligands is of substantial interest [1]. Protein binding influences many pharmacokinetic characteristics and may involve one or several macromolecules. The simplest situation is that in which only one protein is involved. The attraction of a drug toward plasma proteins is an essential concern while defining its complete pharmacokinetic effect [2, 3]. A large extent of protein binding diminishes the free concentration of the drug followed by a consequent decrease in its therapeutic activity. Conversely, circulating protein drug complexes also serve to reload the free drug concentration, as free drug is excreted from the body by various elimination processes, and thus, prolong the duration of drug action. Therefore, the level of protein binding is an important factor in the delicate balance between intended physiological activity and potential side effects of the drug.
Serum albumin is the principal extracellular protein of the circulatory system, and accounts for about 60 % of the total plasma protein and responsible for about 80 % of the colloid osmotic pressure of the blood. Being the major ligand binding and transport protein of circulatory system, it is considered as a model for in vitro study of drug–protein interaction [4]. The primary structure of these transport proteins has about 583–585 amino acid residues and is characterized by a low content of tryptophan and a high content of cystine stabilizing a series of nine loops. The secondary structure of these serum albumins is constituted of 67 % of helix of six turns and 17 disulfide bridges [5–7]. The tertiary structure is composed of three domains, I, II, and III. Each domain is constituted by two subdomains named as IA, IB, IIA, IIB, IIIA, and IIIB [5–7]. Because domains II and III share a common interface, binding of a probe to domain III leads to conformational changes affecting the binding affinities to domain II. Bovine and human serum albumins (BSA and HSA) display approximately 80 % sequence homology and a repeating pattern of disulfides, which are strictly conserved. The molecular weights are 66 kD for BSA and 66.5 kD for HSA. From the spectroscopic point of view, one of the main differences between the two proteins is that BSA has two tryptophan residues (Trp-134 and Trp-212) and HSA has only one (Trp-214). This additional tryptophan residue in BSA is located at position 134, buried in a hydrophobic pocket, and it has been proposed to lie near the surface of the albumin molecule in the second helix of the first domain [7].
Investigating the binding mechanism of the drug with serum albumins is important for the study of toxicology and pharmacokinetics, as it can elucidate the properties of the drug–protein complex and can provide useful information on the structural features that determine the therapeutic effectiveness of the drug [8–11]. Therefore, the studies on this aspect have been an interesting research field in life sciences, chemistry, and clinical medicine [12]. Despite of a large amount of literature on drug-protein binding, unfolding of the proteins by these therapeutic agents has been paid a little attention as the binding started at very low concentration of the drug and unfolding occurs at relatively higher concentrations of the ligands. Several reports on drug-albumin binding also unveiled the partial unfolding of the albumins as a result of the interaction [13–19]. However, most of these studies were performed at very low concentrations of the drugs. We, therefore, have studied the binding as well as the conformational transitions of the bovine serum albumin in presence of several concentrations of sulfadiazine, SD (Scheme 1) by using UV, fluorescence, circular dichroism (CD) and Fourier transform infra-red (FTIR) spectroscopies. In our studies it was found that a typical binding takes place between drug and albumin when the concentration of SD was very low but at higher [SD] unfolding of albumin takes place and the α-helical content of the protein decreases very much with a simultaneous increase in β-sheet conformation. SD is an antibiotic drug which belongs to the sulfonamide category and capable to inhibit the folic acid production inside the bacterial cell causing the elimination of the bacteria [20, 21]. It is used to treat urinary tract infections [22, 23]. Recently it was found that, sulfadiazine was able to partially protect the rat temporal cortex from amyloid beta peptide (25–35)-induced alterations of the somatostatinergic system [24]. During the metabolism of radioactive sulfadiazine in the rat, the concentration of the drug-related material in the plasma or tissues, after 72 h, was less than 0.01 ppm with the exception of the liver (0.13 ppm) [25]. Though, this concentration of SD was found to be in micromoles [25] we have seen the in vitro effect of higher concentrations of SD on the conformation of albumin by taking BSA as a model protein.

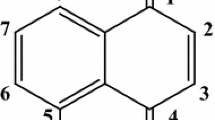
Chemical structure of sulfadiazine
Materials and methods
Materials
Bovine serum albumin (BSA, lyophilized powder, ≥98 %, essentially fatty acid free, #A7030) and Sulfadiazine (SD, 99 %) were purchased from Sigma, USA and used as received.
Procedure
Studies of protein folding are normally carried out in buffered dilute aqueous solutions to avoid loss of protein to the aggregation phenomenon [26]. Stock solutions of BSA (20 mg/ml) and SD, prepared in phosphate buffer of pH 7.4 (well above the isoelectric point of BSA, 4.7, hence the protein possesses a net negative charge at this pH) demineralized double-distilled water of specific conductivity (1–2) × 10−6 s cm−1 was used for preparing the buffer solution. All measurements have been done at 37 °C.
UV measurements
UV spectra, from the range of 240 to 350 nm, were recorded on Perkin-Elmer Lambda 45 Spectrophotometer. Quartz cuvettes of 1 cm path length were used for the measurements. Baseline or reference spectrum was subtracted for each measurement. The absorption titrations were made in such a way that the concentration of the BSA was kept constant with varying concentration of the SD.
Fluorescence measurements
Fluorescence measurements were performed on Hitachi spectrofluorometer (Model F 7000) equipped with a PC. The excitation and emission slits were set at 5 nm and cell with path length of 1 cm was used. The fluorescence spectra were taken with a protein concentration of 0.1 mg/ml. To the 0.1 mg/ml protein solution, requisite volumes of stock additive solutions were added to obtain the samples of desired additive concentration. Intrinsic fluorescence was measured by exciting the protein solution at 295 nm and emission spectra were recorded in the range of 300–450 nm. Synchronous fluorescence spectra were collected at ∆λ = 15 nm and ∆λ = 60 nm.
CD measurements
Circular dichroism measurements were carried out with a Jasco spectropolarimeter, Model J-815, equipped with a microcomputer. The instrument was calibrated with D-10-camphorsulfonic acid and was equipped with a thermostatically controlled cell holder attached to a Neslab RTE-110 water-bath with an accuracy of ±0.1 °C. Spectra were collected with a scan speed of 0.2 nm/min and response time of 1 s. Each spectrum was the average of four scans. The spectra were measured at a protein concentration of 0.1 mg/ml at a path length of 1 cm.
FTIR measurements
Fourier transform infra-red spectra were recorded on a Nicolet 6700 FTIR spectrophotometer (DTGS detector, Ni-chrome source and KBr beamsplitter) from ZnSe windows with a resolution of 16 cm−1 using KBr windows at room temperature. To improve signal to noise ratio, 100 scans were averaged for each spectrum. Spectra were analyzed using the OMNIC software provided with the instrument. Protein concentration of samples was 20 mg/ml.
Results and discussion
UV absorption studies
The absorption spectra of BSA–SD system are shown in Fig. 1. The UV range between 250 and 320 nm (near UV range) is most employed range for the protein structure determinations as all three amino acids (tryptophan, tyrosine and phenylalanine) have prominent absorption bands in this wavelength region. Serum albumins display a UV absorption peak at 280 nm [27]. In presence of SD an increase in intensity (hyperchromic shift) takes place as a result of the interaction between macromolecule and drug. To confirm the interaction between BSA and SD, area under the curve was also calculated for the plots given in Fig. 1. The values for area under curve were found to be 7.16, 1.2 and 10.95 for native BSA, SD and BSA–SD complex, respectively. A significant difference in the area under the curve of native BSA and BSA–SD complex clearly indicate that SD is altering the conformation of BSA. Under our experimental conditions SD have very little absorbance at 280 nm, further, the experimental protein concentration was also very small; therefore, the inner filter effect is negligible [28]. However; we have employed fluorescence spectroscopy to determine various binding and thermodynamic parameters for BSA–SD system.

UV absorption spectra of BSA in the presence of SD [BSA] = 0.1 mg/ml; [SD] = 2 μM
Effect of sulphadiazine on fluorescence of BSA
Fluorescence emission spectra of BSA while excited at 295 nm are given in Fig. 2. It is avowed from the figure that fluorescence intensity decreases on increasing the [SD] as a result of interaction between the drug and macromolecule. These results suggest the dominance of the hydrophobic interaction [29]. Further details of the conformational changes and alterations occur in the fluorophore environment can be obtained by using the synchronous fluorescence spectra of BSA with ∆λ = 60 nm (for Trp) and ∆λ = 15 nm (for Tyr) (vide infra).

Fluorescence emission spectra of BSA (0.1 mg/ml) in presence of various concentrations of SD, curves from 1 to 17 corresponding to SD concentrations of 0, 1, 2.5, 5, 10, 15, 20, 25, 35, 50, 75, 100, 150, 200, 250, 300, 350 μM, respectively when excited at 295 nm
The drug-protein binding information can be obtained from the fluorescence quenching of protein [30, 31]. The following Stern–Volmer equation [32] was used to analyze the fluorescence quenching data (Fig. 3):
where F 0 and F are the steady-state fluorescence intensities in the absence and presence of quencher, respectively, K SV the Stern–Volmer quenching constant, K q stands for bimolecular quenching constant, \({{\uptau}}_{0}\) for the life time of flurophore in the absence of quencher and [Q] is the concentration of quencher (i.e., drug).

Stern-Volmer plot for quenching of BSA fluorescence by SD at 295 nm. Inset: Plot of log (F 0 − F)/F as a function of log [SD]
Since the quenching involved in the drug-albumin binding may be either static (formation of a ground-state complex between the fluorophore and the quencher) or dynamic (phenomenon of collisional encounters between the quencher and fluorophore during the lifetime of the excited state) these can be distinguished by calculating the quenching rate constants, K q which was evaluated by the following equation
The value of \({{\uptau}}_{0}\) for biopolymers is 10−8 s−1 [33]. If the value of the k q is larger than the limiting diffusion rate constant of the biomolecules (2.0 × 1010 L mol−1 s−1), [34, 35] the static quenching could be the main mechanism of the fluorescence quenching otherwise the dynamic quenching occurs.
The modified Stern–Volmer equation used, for the equilibrium between free and bound molecules when small molecules bind independently to a set of equivalent sites on a macromolecule: [36]
where K and n are the binding constant and the number of binding sites, respectively. Thus, a plot of log (F 0 − F)/F versus log [Q] can be used to determine K as well as n (Fig. 3, inset).
The intercept and slope of the plot of log (F 0 − F)/F versus log [Q] were used to calculate the values of K & n for drug–serum albumin systems.
The binding constant K was used to calculate the standard free energy change ∆Gº of the ligand binding to the serum albumins from the relationship.
The values of K, n & ∆G°binding are summarized in Table 1 and the value of K, is significant to understand the distribution of the drug in plasma since the weak binding can lead to a short lifetime or poor distribution, while strong binding can decrease the concentration of free drug in plasma.
The results have been analyzed according to Eqs. (1) and (3). The values of K SV and K q are listed in Table 1. For the fluorometric titration of the SD into BSA solution, the Stern–Volmer quenching constant, K SV = 4.24 × 104 mol dm−3, was calculated from the good linear relationship between F 0 /F and [SD]. K q was calculated from the relationship given in Eq. 2 while τ 0 was taken as 10−8 s involvement of the static quenching in the binding process was concluded on the basis of calculate value of K q [34, 35].
An alternative method, the Lineweaver–Burk method, was also used to analyze the quenching data. This method is regarded as more prominent while describing static quenching [37–40] rather Stern–Volmer method, which describes dynamic quenching [41–43]. The Lineweaver–Burk equation is
where K LB is the static quenching constant (M−1), which describes the binding efficiency of small ligands to the macromolecules at ground state [42]. K LB calculated from the linear relation (Fig. 4) of Eq. (5) is given in Table 1 and is of the order of the calculated value of K SV . From the comparison of linear regression of Fig. 3 and Fig. 4 it can be concluded that Lineweaver–Burk curve has better linear relation as compared to the Stern–Volmer curves which confirms that the quenching is static [27]. Similarly higher value of K LB as compared to the K SV also confirms this. As observed with our UV absorption spectra (Fig. 1), there is some perturbation in the absorption spectrum of BSA in presence of SD. This is also a proof of the static quenching as collisional quenching only affects the excited states of the fluorophores, and thus no change in absorption spectra should be observed. In contrast, ground-state complex formation will frequently result is perturbation of the absorption spectrum of the fluorophore [32]. Therefore, in our case, it can be concluded that the binding of SD to BSA is governed by static quenching through the formation of the drug-protein complex.

Lineweaver-Burk plot of BSA–SD interaction
Synchronous fluorescence spectroscopy
The synchronous spectra are a characteristic of the typical Trp residue fluorescence at ∆λ (λem−λex) = 60 nm, while they are characteristic of the typical Tyr residue fluorescence at ∆λ = 15 nm [44]. Figures 5a and 6b display the effect of [SD] on the synchronous spectra of BSA. A large red shift in the wavelength maximum was observed in both cases when the concentration of the SD increased from 0 to 350 μM. The observed results indicate that the polarity surrounding all Trp as well as Tyr residues is changing on increasing the drug concentration as a result of the drug-polymer interaction. However, from the spectra observed at low concentrations of the drug, it was found that despite a decrease in the fluorescence intensity (as a result of quenching) the wavelength of absorption maximum show very small difference (up to 20 mM the change was <1 whereas from 20 to 50 nm the decrease of about 1 nm in both cases (i.e., ∆λ = 15 and 60) was observed) which reveals that the conformation of the albumin was remained unchanged up to 20 mM SD followed by a small perturbation in the range of 20–100 mM of SD. The initial constancy in λmax with a decrease in fluorescence intensity up to 20 μM of SD is suggestive of the simple binding of the drug to the albumin whereas from 20 to 100 mM binding also results in a small change of the conformation of BSA. [SD] ≥150 mM was even more capable to unfold the protein as unveiled by a significant red shift of 9 nm in case of ∆λ15 and 6 nm for ∆λ60 which signify the role of SD to disrupt the secondary structure of the protein. This observed red shift indicates that hydrophobicity around these residues (Trp and Tyr) is decreasing and this may be probably due to the dipole moment created by the sulfonamide group present in SD. These outcomes are in consistent with the results obtained from the CD spectrophotometry.

Synchronous fluorescence spectra of BSA–SD system a ∆λ = 15 b ∆λ = 60. curves from 1 to 17 corresponding to SD concentrations of 0, 1, 2.5, 5, 10, 15, 20, 25, 35, 50, 75, 100, 150, 200, 250, 300, 350 μM and [BSA] = 0.1 mg/ml

Selected circular dichroism spectra of BSA–SD system: curves from 1 to 7 corresponding to SD concentrations of 0, 1, 10, 25, 50, 75, 150 μM and [BSA] = 0.1 mg/ml
Circular dichroism spectroscopy
Circular dichroism was employed to ascertain the possible influence of SD binding on the secondary structure of the BSA [45]. α-helices exhibit immense CD absorption with negative ellipticity at 222 and 208 nm and positive ellipticity at 193 nm. Modifications of ellipticity at 222 nm (−MRE222) and 208 nm (−MRE208) are convenient methods for observing changes in α-helical content [46, 47]. Several CD spectra in presence and absence of various concentrations of [SD] are given in Fig. 6.
The CD in millidegree, obtained over the wavelength range of 200–250 nm, was converted to mean residue ellipticity (MRE, θ) using the following conversion:
where θ obs is the CD in millidegree, n is the number of amino acid residues (585), l is the cell-path length in cm and C p the molarity. The unit of MRE is deg cm2 dmol−1.
The secondary structure was estimated from spectra between 200 and 240 nm using K2D3 CD secondary structure server, which uses an unsupervised neural network to predict secondary structure [48].
The α-helical content of pure BSA was found to be 62.2 % that is very close to the literature value (Table 2) [45]. Addition of small amounts of SD (0–20 mM) into BSA solution caused small difference in the % α-helix of BSA (Table 2), however, from 25 to 100 mM SD a considerable change in the α-helical content was observed followed by an almost disruption of α-helix from 150 to 300 mM of SD. An increase in β-sheets was also observed on increasing the concentration of drug and it dominated the α-helical content at higher experimental concentrations of SD. The results are in accordance with the one observed with synchronous fluorescence spectroscopy.
FTIR spectroscopy
Infrared spectroscopy is a very powerful tool for studying the secondary structure of the proteins and their dynamics [49–51]. The assessment of the frequencies at which the amide bonds absorb the radiation are useful probes to evaluate the secondary structure. The amide bands I which is 80 % C=O stretch occurs near 1650 cm−1 though amide band II, due to C–N and N–H stretch arises near 1550 cm−1. Amide I band is considered more sensitive as compared to the amide II band for estimating the secondary structure of the protein. After analyzing CD and synchronous spectroscopic results of the present study, we have selected 50 mM of drug to see the effect of SD on secondary structure of BSA. The FTIR difference spectra of BSA and BSA–SD system are given in Fig. 7. The peak positions of both amide bands shifted in presence of SD. The peak position of amide I band shifted from 1656 to 1650 cm−1 which signifies a large change in the secondary structure of BSA. Similarly the peak position of amide II band is also shifted from 1542 to 1541. The SD has interacted with C=O group present in polypeptide chains which results in the rearrangement of polypeptide carbonyl hydrogen bonding network.

FTIR spectra and difference spectra of BSA in aqueous buffered solution. a FTIR spectrum of BSA (20 mg/ml), b FTIR difference spectrum of BSA obtained by subtracting the spectrum of the SD-free from that of the BSA and SD bound form in the region of 1700–1500 cm−1 ([SD] = 500 μM)
Three-dimensional fluorescence spectra of BSA–SD system
To get more insight on the binding of BSA and SD, three-dimensional fluorescence spectroscopy was performed on BSA and BSA–SD complex. The three-dimensional spectra of BSA–SD system under various conditions of SD are given in Fig. 8 and the fluorescence spectral peak position, intensity and Stokes shift are summarized in Table 3. The three-dimensional spectroscopy is a growing analysis technique in the present time and can provide more detailed information about the protein conformation. The contour map displayed a bird’s eye view of the fluorescence spectra. Peak 1 is the Rayleigh scattering peat at which excitation wavelength equals to the emission wavelength while peak 2 mainly discloses the spectral characteristics of Trp and Tyr residues and peak 3 may mainly exhibit the fluorescence characteristic of polypeptide backbone [51, 52]. Addition of SD causes no diversification in the position of excitation and emission wavelengths and the stokes shift. But the molecular microenvironment in the vicinity of the Trp residue of BSA was a distinct change in the polarity after conjugation of SD with BSA. In fact, the analysis of Trp excitation and emission spectra can provide subservient information about the changes of the protein conformational state and Trp microenvironment, these data being closely related to its solvent exposure and hydrophobicity. Our observations reveal that the Trp residue of BSA was accompanied by a decrease in the fluorescence intensity, peak ratio 2.21:1.87:1.00 in the absence and presence of SD. The above phenomenon and analysis of fluorescence characteristics of the peaks revealed that a complex between BSA and SD has formed and binding induced some microenvironmental and conformational changes in BSA. These conclusions are found in a good agreement with the findings observed by CD and synchronous fluorescence spectroscopies.

The three dimensional fluorescence spectral diagrams of BSA (0.1 mg/ml) in absence (a) and presence of b 25 μM and c 100 μM SD
Energy transfer from BSA to SD
Förster’s nonradiative energy transfer theory is a distance dependent interaction in which excitation energy is transferred nonradiatively form donor to acceptor. There is a spectral overlap between the fluorescence emission spectra of free BSA and absorption spectrum of SD (Fig. 9). According to FRET [53] energy transfer is likely to happen under the following conditions: (i) the relative orientation of the donor and acceptor dipoles (ii) the extent of overlap of fluorescence emission spectrum of the donor with the absorption spectrum of the acceptor, and (iii) the distance between the donor and the acceptor is less than 8 nm. Based on Förster’s theory, the efficiency of energy transfer (E) is related to the distance r between donor and acceptor by Eq. (7) [32].
where r is the distance between acceptor (SD) and donor (BSA) and R 0 is the critical distance when the transfer efficiency is 50 %. The value for R 0 is calculated using Eqs. (7) and (8): [32].

Spectral overlap of SD absorption (curve A) with fluorescence of BSA. Concentrations of BSA and SD were 1.5 × 10−6 and 2.5 × 10−6 M, respectively
where N is the refractive index of the medium, k2 is the orientation factor, and Φ is the quantum yield of the donor. The spectral overlap integral (J) between the donor emission spectrum and the acceptor absorbance spectrum was approximated by the following summation
where F(λ) and ε(λ) represent the fluorescence intensity of the donor and the molar extinction coefficient of the acceptor, respectively. From these relationships J, E and R 0 can be calculated. For the BSA–ligand interaction, K 2 = 2/3, N = 1.336 and Φ = 0.15 [43]. According to the above equations following values of the parameters were obtained: J = 3.0 × 10−16 cm3 L mol−1, R 0 = 1.42 nm, E = 0.142, and r = 1.92 nm. As the donor–to-acceptor distance for the BSA–SD system is less than 8 nm and 0.5 R 0 < r<1.5 R 0, which implies high probability of energy transfer from BSA to SD. Besides, the donor-to-acceptor distance is less than 8 nm, indicating again that the static quenching interaction occurred between BSA and SD.
Conclusions
Interaction of bovine serum albumin (BSA) with an antibiotic sulfonamide drug, sulfadiazine (SD) was studied. Results showed that there was a strong (1:1) binding, which involved static quenching, between the SD and BSA with very small effect on the conformation of protein at low concentration of drug. However, in presence of high concentration of SD a complete disruption of α-helical content was observed with a significant increase in β-sheet conformations. Dominance of the hydrophobic interaction was found to be involved in the drug-albumin binding. In addition, a high probability of energy transfer from BSA to SD was also observed.
References
Vuignier K, Schappler J, Veuthey J-L, Carrupt P-A, Martel S (2010) Drug–protein binding: a critical review of analytical tools. Anal Bioanal Chem 398:53–66
Florence AT, Attwood D (2011) Physicochemical principles of pharmacy. Pharmaceutical Press, London
DiPiro JT, Spruill WJ, Wade WE, Blouin RA, Pruemer JM (2010) Concepts in clinical pharmacokinetics, 5th edn. American Society of Health Systems Pharmacist, Bethesda
Watanabe Y, Hayashi T, Kitayama R, Yasuda T, Saikawa I, Shimizu K (1981) Studies on protein binding of antibiotics. II. Effect of apalcillin on protein binding and pharmacokinetics of cefoperazone and cefazolin. J Antibiot (Tokyo) 34:753–757
Carter DC, Ho JX (1994) Structure of serum albumin. Adv Protein Chem 45:153–203
Min HX, Carter DC (1992) Atomic structure and chemistry of human serum albumin. Nature 358:209–215
Peters T (1985) Serum albumin. Adv Protein Chem 37:161–245
Abou-Zied OK, Al-Lawatia N, Elstner M, Steinbrecher TB (2013) Binding of hydroxyquinoline probes to human serum albumin: combining molecular modeling and Förster’s resonance energy transfer spectroscopy to understand flexible ligand binding. J Phys Chem B 117:1062–1074
Tardioli S, Lammers I, Hooijschuur J-H, Ariese F, van der Zwan G, Gooijer C (2012) Complementary fluorescence and phosphorescence study of the interaction of brompheniramine with human serum albumin. J Phys Chem B 116:7033–7039
Tardioli S, Buijs J, Gooijer C, van der Zwan G (2012) pH-Dependent complexation of histamine H1 receptor antagonists and human serum albumin studied by UV resonance Raman spectroscopy. J Phys Chem B 116:3808–3815
Nuin E, Andreu I, Torres MJ, Jimenez MC, Miranda MA (2011) Enhanced photosafety of cinacalcet upon complexation with serum albumin. J Phys Chem B 115:1158–1164
Burton ME, Shaw LM, Schentag JJ, Evans WE (2006) Applied pharmacokinetics & pharmacodynamics: principles of therapeutic drug monitoring. Lippincott Williams & Wilkins, Baltimore, MD
Bourassa P, Dubeau S, Maharvi GM, Fauq AH, Thomas TJ, Tajmir-Riahi HA (2011) Binding of antitumor tamoxifen and its metabolites 4-hydroxytamoxifen and endoxifen to human serum albumin. Biochimie 93:1089–1101
Galantini L, Leggio C, Konarev PV, Pavel NV (2010) Human serum albumin binding ibuprofen: a 3D description of the unfolding pathway in urea. Biophys Chem 147:111–122
Jurasekova J, Marconi G, Sanchez-Cortes S, Torregiani A (2009) Spectroscopic and molecular modeling studies on the binding of the flavonoid luteolin and human serum albumin. Biopolymer 91:917–927
Liu Y-C, Yang Z-Y, Du J, Yao X-J, Lei R-X, Zhong X-D, Liu J-N, Lu H-S, Li H (2008) Study on the interactions of kaempferol and quercetin with intravenous immunoglobulin by fluorescence quenching, Fourier transformation infrared spectroscopy and circular dichroism spectroscopy. Chem Pharm Bull 56:443–451
Bourassa P, Dubeau S, Maharvi GM, Fauq AH, Thomas TJ, Tajmir-Riahi HA (2011) Locating the binding sites of anticancer tamoxifen and its metabolites 4-hydroxytamoxifen and endoxifen on bovine serum albumin. Eur J Med Chem 46:4344–4353
Liu Y, Yang Z, Du J, Yao X, Lui R, Zhang X, Lui J, Hu H, Li H (2008) Interaction of curcumin with intravenous immunoglobulin: a fluorescence quenching and Fourier transformation infrared spectroscopy study. Immunobiology 213:651–661
Liu Y, Yang Z, Du J, Yao X, Lui R, Zhang X, Lui J, Hu H, Li H (2008) Interaction of Taxol with intravenous immunoglobulin: an inhibition of Taxol from crystallizing in aqueous solution. Int Immunopharmacol 8:390–400
Brown GM (1962) The biosynthesis of folic acid. II. Inhibition by sulfonamides. J Biol Chem 237:536–540
Kleineidam K, Sharma S, Kotzerke A, Heuer H, Thiele-Bruhn S, Smalla K, Wilke B-M, Schloter M (2010) Effect of sulfadiazine on abundance and diversity of denitrifying bacteria by determining nirK and nirS genes in two arable soils. Microb Ecol 60:703–707
Hanson E, Hansson S, Jodel U (1989) Trimethoprim-sulphadiazine prophylaxis in children with vesico-ureteric reflux. Scandinavian J Infect Dis 21:201–204
Makela P, Kalima S, Gordin A, Sundberg S, Ojajarvi P, Mannisto PT (1983) Pipemidic acid and sulfadiazine in acute urinary tract infections. Curr Therap Res 34:757–761
Burgos-Ramos E, Puebla-Jimenez L, Hernandez-Pinto A, Arilla-Ferreiro E (2009) Sulfadiazine partially protects the rat temporal cortex from amyloid beta peptide (25-35)-induced alterations of the somatostatinergic system. Neuroendocrinology 89:400–410
Woolley JL, Sigel CW (1979) Metabolism and disposition by the rat of 35S-sulfadiazine alone and in the presence of trimethoprim. Drug Metab Dispos 7:94–99
Byrne N, Wang L-M, Belieres J-P, Angell C A (2007) Reversible folding–unfolding, aggregation protection, and multi-year stabilization, in high concentration protein solutions using ionic liquids. Chem Commun 2714–2716. DOI: 10, 1039/b618943a
Mach H, Volkin DB, Burke CJ, Middaugh CR (1995) Ultraviolet absorption spectroscopy. In: Shirley BA (ed) Protein stability and folding: theory and practice, methods in molecular biology, vol 40. Humana Press, Totowa, pp 91–114
Zhang H, Huang X, Zhang M (2008) Spectral diagnostics of the interaction between pyridoxine hydrochloride and bovine serum albumin in vitro. Mol Biol Rep 35:699–705
Mandeville JS, Froshlich E, Tajmir-Riahi HA (2009) Study of curcumin and genistein interactions with human serum albumin. J Pharm Biomed Anal 49:468–474
Li Y, He W, Liu J, Sheng F, Hu Z, Chen X (2005) Binding of the bioactive component jatrorrhizine to human serum albumin. Biochim Biophys Acta 1722:15–21
Tian J, Liu J, Chen X (2005) Interaction of wogonin with bovine serum albumin. Bioorg Med Chem 13:4124–4129
Lakowicz JR (1999) Principles of Fluorescence Spectroscopy, 3rd edn. Springer, New York
Togashi DM, Ryder AG (2008) A fluorescence analysis of ANS bound to bovine serum albumin: binding properties revisited by using energy transfer. J Fluorescence 18:519–526
Eftink MR (1991) Fluorescence quenching reactions: probing biological macromolecular structure. In: Dewey TG (ed) Biophysical and biochemical aspects of fluorescence spectroscopy. Plenum, New York, pp 1–41
Ware WR (1962) Oxygen quenching of fluorescence in solution: an experimental study of the diffusion process. J Phys Chem 66:455–458
Kang J, Lu Y, Xie MX, Li S, Jiang M, Wang YD (2004) Interactions of human serum albumin with chlorogenic acid and ferulic acid. Biochim Biophys Acta 1674:205–214
Zhang H, Huang X, Zhang M (2008) Spectral diagnostics of the interaction between pyridoxine hydrochloride and bovine serum albumin in vitro. Mol Biol Rep 35:699–705
Jiang C-Q, Gao M-X, Meng X-Z (2003) Study of the interaction between daunorubicin and human serum albumin and the determination of daunorubicin in blood serum samples. Spectrochim Acta Part A 59:1605–1610
Darwish S, Abu-Sharkh S, Abu-Tier M, Makharza S, Abu-Hadid M (2010) Spectroscopic investigations of pentobarbital interaction with human serum albumin. J Mol Str 963:122–129
Ding F, Liu W, Zhang X, Wu L, Zhang L, Sun Y (2010) Identification of pyrazosulfuron-ethyl binding affinity and binding site subdomain IIA in human serum albumin by spectroscopic methods. Spectrochim Acta Part A 75:1088–1094
Bian H, Li M, Yu Q, Chen Z, Tian J, Liang H (2006) Study of the interaction of artemisinin with bovine serum albumin. Int J Biol Macromol 39:291–297
Zhang H-X, Huang X, Mei P, Li K-H, Yan C-N (2006) Studies on the interaction of tricyclazole with β-cyclodextrin and human serum albumin by spectroscopy. J Fluoresc 16:287–294
Cui F-L, Fan J, Li J-P, Hu Z-D (2004) Interactions between 1-benzoyl-4-p-chlorophenyl thiosemicarbazide and serum albumin: investigation by fluorescence spectroscopy. Bioorg Med Chem 12:151–157
Veskin NL (2002) Photonics of biopolymers, 2nd edn. Springer, Heidelberg
Peters T Jr (1995) All about albumin: biochemistry, genetics, and medical applications. Academic Press, London
Kelly SM, Jess TJ, Price NC (2005) How to study proteins by circular dichroism. Biochim Biophys Acta 1751:119–139
Kelly SM, Price NC (2000) The use of circular dichroism in the investigation of protein structure and function. Curr Protein Pept Sci 1:349–384
Louis-Jeune C, Andrade-Navarro MA, Perez-Iratxeta C (2012) Prediction of protein secondary structure from circular dichroism using theoretically derived spectra. Proteins Struc Func Bioinfo 80:374–381
Cheng Z (2012) Comparative studies on the interactions of honokiol and magnolol with human serum albumin. J Pharm Biomed Anal 66:240–251
Bourassa P, Tajmir-Riahi HA (2012) Locating the binding sites of folic acid with milk α- and β-caseins. J Phys Chem B 116:513–519
Shen H, Gu Z, Jian K, Qi J (2013) In vitro study on the binding of gemcitabine to bovine serum albumin. J Pharm Biomed Anal 75:6–93
Suryawanshi VD, Anbhule PV, Gore AH, Patil SR, Kolekar GB (2012) Spectroscopic investigation on the interaction of pyrimidine derivative, 2-amino-6-hydroxy-4-(3,4-dimethoxyphenyl)-pyrimidine-5-carbonitrile with human serum albumin: mechanistic and conformational study. Ind Eng Chem Res 51:95–102
Förster T (1965) Delocalized excitation and excitation transfer. In: Sinanoglu O (ed) Modern quantum chemistry. Academic Press, New York, pp 93–137
Acknowledgments
This Project was supported by King Saud University, Deanship of Scientific Research, College of Science Research Center.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ali, M.S., Al-Lohedan, H.A. Sulfadiazine binds and unfolds bovine serum albumin: an in vitro study. Mol Biol Rep 40, 6081–6090 (2013). https://doi.org/10.1007/s11033-013-2719-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-013-2719-8