
Abstract
Because of its lower toxicity and good tolerability and response, gemcitabine has been described as one of the most highly promising drugs for urinary bladder cancer therapy. Its phosphorylated active-dFdCTP metabolite can incorporate into DNA, causing replication blockage. Additionally, it is known that mutations in the TP53 gene are related to the high recurrence rate of these neoplasias. Based on these premises, we investigated the effects of gemcitabine on the expression of the cell cycle-related genes in two different TP53-mutated bladder transitional carcinoma cell lines–5637 (from a moderate-grade tumor with a TP53 allele carrying two mutations) and T24 (from an invasive tumor with a TP53 allele encoding an in-frame deletion). Cell viability and morphology analyses (phase-contrast photomicrographs), Nuclear Division Index and pathway-specific quantitative RT-PCR gene arrays were performed. Treatment with gemcitabine led to the following results: (1) a significant decrease of viable T24 cells after treatment at the highest concentration (3.12 μM) tested; (2) scattered, elongated and vacuolated 5637 and T24 cells; (3) a cytostatic effect in both cell lines; and (4) significant upregulation of the BRCA1, CCNE1, CDK2, CDK6, CDKN1A, CDKN2B, E2F4, GADD45A, MAD2L2, CCNH, SERTAD1, CDC1, and CHEK1 genes. Gemcitabine had distinct toxicogenomic effects in the bladder transitional carcinoma cell lines with two different TP53 mutations. However, independent of the type of mutation and tumor grade, gemcitabine induced cell cycle arrest; upregulation of DNA repair-related genes, G1/S transition, apoptosis and activation of transcription factors, mainly by upregulation of the CCNE1, CDKN1A and GADD45A genes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In most countries of the Western world, bladder cancer is predominantly of the transitional cell carcinoma (TCC), which comprises 90 % of all bladder carcinomas, while 5 % of them are identified as squamous and 2 % as adenocarcinomas [1]. Although men are 3–4 times more likely to develop bladder cancer, women present more often with advanced disease and have a lower chance of survival [2]. About 30 % of TCCs display solid and invasive growth patterns, being locally advanced or metastatic at the time of diagnosis; the other 70 % are confined to the epithelium or subepithelial connective tissue [3]. Urothelial bladder cancers have been reported to have the highest recurrence rate among solid tumors. Their recurrence ranges from 50 % to 70 %, while more than 15 % progress to muscle invasion over a 5-year period [4]. Among these, mutations in the TP53 gene are the most important alterations frequently found in bladder cancer cells [5].
Chemotherapeutic protocols have been studied extensively with the hope of improving overall survival and treating the cancer. It is known that the characterization of genes associated with tumor sensitivity to antineoplastic agents can play a critical role in the selection of preferable treatments [6]. Based on its lower toxicity and good tolerability and response, when compared to several chemotherapeutic drugs, gemcitabine has shown promising results when tested alone [7]. In 2006, Bellmut [8] described that gemcitabine is the single most highly promising agent for bladder cancer. Furthermore, clinical data have also indicated that this drug has activity in terms of tumor response and overall survival [9]. Gemcitabine is a deoxycytidine analog, which is phosphorylated to an active dFdCTP metabolite (gemcitabine triphosphate) that incorporates into DNA, causing replication blockage [10], and also into RNA, inhibiting RNA synthesis [11]. Gemcitabine has a molecular weight of 299 Da, which is lower than those of the commonly used intravesical chemotherapeutic agents, such as mitomycin C and doxorubicin. This may enable gemcitabine to penetrate the bladder mucosa with beneficial effects in the treatment of invasive bladder cancers [12].
After incorporation into the cell, gemcitabine can be recognized by the p53 protein [13]. In wild-type TP53 cells, gemcitabine-induced cellular damage can stimulate p53 expression, resulting in p21 expression and cell cycle arrest, enabling the cell to repair the DNA damage or inducing apoptosis mediated by the BAX gene. In cells with a mutated TP53 phenotype, p53 and p21 cannot be induced, but BAX expression can still be found, resulting in apoptosis [14]. Additionally, gemcitabine can inhibit DNA synthesis by causing DNA strand breaks, thereby eliciting a DNA damage response characterized by cell cycle arrest in the G1/S phase [15]. In fact, da Silva et al. [16] have shown that gemcitabine induces G1-phase arrest and triggers apoptosis in bladder cancer cell lines. To explain this mechanism, Toshimitsu et al. [17] have shown that in pancreatic cancer cells, gemcitabine upregulated the gene, a cyclin-dependent kinase inhibitor implicated in the transition from proliferation to a quiescent state. These authors suggest that the elevated expression of CDKN1B might cause G1 arrest of cells. Another possible mechanism would include the inhibition of DNA topoisomerases [18]. Therefore, these targets could represent opportunities for chemotherapeutic intervention, contributing to the treatment of cancer [19]. In this direction, high-throughput methods may help to better understand the molecular activities in different cell types, promoting the understanding of the mechanism of action of these drugs [20].
Therefore, based on the fact that gemcitabine is a promising therapy for different types of cancer, because its mechanism of action is not well-known, and because of the importance of TP53 mutations for bladder cancer, we investigated the effects of gemcitabine on bladder cancer cells carrying different TP53 mutations. The expression of cell cycle-related genes was analysed. Thus, we evaluated whether gemcitabine activity would be dependent on the TP53 genetic background of tumor cells.
Materials and methods
Cell lines
The two human bladder TCC cell lines, 5637, with a TP53 allele carrying two mutations (one at codon 280—Arg>Thr and the other at codon 72—Arg>Pro), obtained from a moderate grade tumor and T24, with a TP53 allele encoding an in-frame deletion of tyrosine 126, obtained from an invasive tumor, were acquired from the Cell Bank of the Federal University of Rio de Janeiro, Brazil. The cells were maintained as previously described by da Silva et al. [16]. The antineoplastic drug gemcitabine (dFdC, Gemzar) was obtained from Eli Lilly Laboratory (USA). Ultra-pure sterilized water was used for the dilutions.
Experimental design
The cells were seeded into 12-well culture plates (1 × 104 cells/well) for evaluating the cell viability and the morphological changes, into Petri dishes (1 × 106/cells/dish) for evaluating the Nuclear Division Index (NDI) and into 25 cm3 culture flasks (2 × 106 cells/flask) for the RNA extraction. Twenty-four hours after seeding (T1 = 0 h), the cells were treated with gemcitabine at concentrations of 0.78, 1.56, or 3.12 μM (as defined in previous experiments) [16], during a 24 hour period (T2 = 24 h). Untreated cells were cultured in parallel as a negative control. During T2, the cells were washed with Hank’s solution (0.4 g KCl, 0.06 g KH2PO4, 0.04 g Na2HPO4, 0.35 g NaHCO3, 1 g glucose and 8 g NaCl in 1,000 ml H2O) and collected for morphological, cell viability and gene expression evaluations.
Cell viability, morphology and NDI
Cell viability was evaluated by the trypan blue exclusion test. The assay was performed in triplicate, and the results were represented as the mean ± standard deviation (SD). A phase-contrast microscope was used before and after the gemcitabine treatment for evaluating the morphological changes.
For the NDI assay, the 5637 and T24 cell lines were treated with gemcitabine at concentrations of 0.78, 1.56, or 3.12 μM during a 6-h period. Afterwards, the cells were washed, and cytochalasin B (Sigma-Aldrich) at final concentration of 3 μg/ml was added. Twenty-four hours later, the cells were collected, the suspension was transferred to 15 ml centrifuge tubes and 5 ml of a hypotonic solution (KCl 0.075 M) was added. After a 5 min centrifugation at 1,000 rpm, the cells were fixed with 5 ml of a methanol:acetic acid (5:1) solution plus 0.3 ml of formaldehyde, and the slides were prepared. The slides were stained with a 5 % Giemsa solution and scored under a light microscope at 400× magnification (2,000 cells/treatment). Cells treated with doxorubicin, at a concentration of 0.4 μg/ml for 2 h, were used as a positive control. The NDI was determined as previously described by Fenech et al. [21]: NDI = M1 + 2 (M2) + 3 (M3) + 4 (M4)/N, where M1–M4 is the number of cells with 1–4 nuclei and N is the total number of viable cells. The assay was performed in duplicate.
RNA extraction
Total RNA was extracted using the Mini RNeasy kit (Qiagen) according to the manufacturer’s instructions. The extracted RNA was stored at −80 °C. The integrity and quality of the RNA was evaluated with 2 % denaturing agarose gels and NanoVue equipment (GE Healthcare), respectively.
PCR arrays
For the gene expression evaluation, gemcitabine was used at a concentration of 1.56 μM (this concentration is genotoxic in the comet assay, but not cytotoxic in the trypan blue and XTT tests [16]. The cell cycle pathway PCR Array (PAHS-020A, SA Biosciences) was used for the qRT-PCR. The cDNA was produced using the RT2 First Strand kit (SA Biosciences) according to manufacturer. An aliquot of the diluted first-strand synthesis reaction was added to the SYBR Green/ROX master mix (SA Biosciences) along with nuclease free water according to the PCR array system’s user manual. Afterwards, 25 μl of the cDNA/master mix cocktail was placed into each well of the pathway-specific qRT-PCR microplate. Quality controls for the genomic DNA contamination, reverse transcription efficiency, and PCR amplification efficiency were analyzed. The qRT-PCR array data were normalized using the arithmetic mean of five housekeeping genes B2M, HPRT1, RPL13A, GAPDH and ACTB for the T24 cells and GAPDH and HPRT1 for the 5637 cells. The arrays were all performed in triplicate. The information regarding the biological functions was obtained from FATIGO (http://babelomics.bioinfo.cipf.es/).
Statistical analysis
For statistical analysis of the NDI and cell viability, a one-way ANOVA test was used. For the gene expression analysis, the fold change was used. In this case, the p value was calculated with Student’s t test with triplicate values for each gene in the control and treatment groups. A p value <0.05 was considered statistically significant.
Results
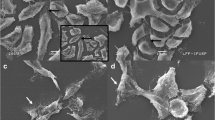
The percentages of viable cells in the 5637 cell line after the treatments with gemcitabine at concentrations of 0, 0.78, 1.56, and 3.12 μM were 94.26 ± 2.27, 86.53 ± 12.89, 76.90 ± 15.13, and 77.96 ± 9.82, respectively; for the T24 cells, the percentages were 98.33 ± 2.88, 97.10 ± 5.02, 89.73 ± 3.06, and 86.23 ± 2.76, respectively. A statistically significant decrease in the percentage of viable cells was only detected in the T24 lineage treated with the highest concentration of gemcitabine (3.12 μM). With regard to the NDI, a statistically significant decrease in the number of cells (p < 0.05) was observed in both cell lines after treatment with the three concentrations of gemcitabine (Table 1). The phase-contrast photomicrographs of the 5637 and T24 cell lines showed scattered, elongated and vacuolated cells after the gemcitabine treatment. Additionally, a lower number of cells were observed in the gemcitabine-treated cell cultures than in their respective controls (Fig. 1).

Photomicrography of two lineages of human bladder transitional carcinoma cells before and after gemcitabine treatment: a 5637 cells; b 5637 cells after treatment with 1.56 μM gemcitabine; c T24 cells; d T24 cells after treatment with 1.56 μM gemcitabine. The arrows show the elongated and vacuolated cells. Phase-contrast microscope, ×400
Of the 84 genes analyzed, 36 genes in the T24 and 29 genes in the 5637 cells were differentially modulated after the gemcitabine treatment (Tables 2 and 3). The highest changes (fold change >2, p < 0.05) were detected for the BIRC5, BRCA1, CCNE1, CDK2, CDK6, CDK8, CDKN1A, CDKN2B, CUL2, E2F4, GADD45A, HERC5, KPNA2, MAD2L1, MAD2L2, NBN, RAD51, RB1 and TFDP2 genes in the 5637 cells and for the BRCA2, CCNE1, CDKN1A, GADD45A, TP53, CCNB1 and CCNF genes in the T24 cells. Therefore, the genes that were equally modulated in both cell lines were CCNE1, CDKN1A and GADD45A (fold change >2, p < 0.05). Other genes (BRCA1, CDK2, CDK6, CDKN2B, E2F4, MAD2L2, CCNH, SERTAD1, CDC16, CHEK1) presented a significant upregulation in the two cell lines (p < 0.05), but the fold change for one or both cell lines was less than two (Figs. 2 and 3).

Scatter plots representing all of the significantly modulated genes in the T24 and 5637 cells, respectively. Each dot represents one gene; the dots above the superior and below the inferior lines represent the highest changes of gene expression, fold change >2 or <2, respectively

Venn diagram showing the upregulated genes in both the T24 and 5637 cell lines
Discussion
It is known that mutations in the TP53 gene are related to the high recurrence rate of urinary bladder cancers [5], and it has been reported that gemcitabine is one of the most highly promising drugs for the treatment of these neoplasias [8]. Therefore, based on these premises, we investigated the effects of gemcitabine on the expression of the cell cycle-related genes in two different TP53-mutated bladder transitional carcinoma cell lines. Previously, we have observed that gemcitabine had similar effects in 5637 and T24 cells, i.e., G1-phase arrest and late apoptosis were visualized in both cell lines [16]. Nevertheless, we did not identify which genes might be involved in these pathways. In fact, similar findings had been reported by Fencher et al. [22], showing that gemcitabine-induced apoptosis in bladder cancer cells is independent of the TP53 status.
Before analyzing the gene expression signature, we checked the effects of gemcitabine on the cell viability and morphology, and on the NDI. The only significant difference detected between the two cell lines was a decreased viability of the T24 cells just after the exposure to the highest concentration of gemcitabine (3.12 μM). The observed morphological changes (vacuolized, scattered and elongated cells) together with a low cell density in both lineages were suggestive of cell cycle arrest and late apoptosis. The reduced NDIs confirmed that the blockage of cell division is independent of the gemcitabine concentration used. According to Fenech [21], the NDI indicates the proliferation state of the viable cells. Therefore, it can be used as a marker of cytostatic effect. Aydemir et al. [23] also found a decreased rate of cell replication in cultured human lymphocytes after gemcitabine treatment. A possible mechanism to explain this effect is the ability of gemcitabine to incorporate into the DNA strands and inhibit the DNA polymerase activity [24].
In general, toxicogenomic research focuses on the modulation of the gene expression profiling or pathways after the exposure to a toxic agent. If the concentration of an agent induces significant levels of cell death, the transcriptome profiles will be reflective of the dead or dying cells instead of the cellular responses to a lower non-cytotoxic concentration [25]. Therefore, the concentration we used for evaluating the toxicogenomic effect of gemcitabine (1.56 μM) was carefully selected based on the genotoxic (data not shown) and cytotoxic data previously published by our group [16]. The gene arrays initially showed upregulation of the BRCA1, CCNE1, CDK2, CDK6, CDKN1A, CDKN2B, E2F4, GADD45A, MAD2L2, CCNH, SERTAD1, CDC16, and CHEK1 genes in the 5637 and T24 cells after gemcitabine treatment. According to the gene ontology (GO), these genes are primarily involved in the negative regulation of the cell cycle (BRCA1, CDK2, CDK6, CDKN1A, CDKN2B, GADD45A, MAD2L2, CDC1, and CHEK1), cell cycle arrest (BRCA1, CDK2, CDKN1A, CDKN2B, GADD45A, MAD2L2, CDC16, and CHEK1), G1/S transition of the mitotic cell cycle (CCNE1, CDK2, CDKN1A, CDKN2B, E2F4, and CCNH), DNA repair (BRCA1 and GADD45A), apoptosis (BRCA1 and GADD45A) and the regulation of transcription (SERTAD1 and E2F4). Several authors have shown that some of these genes can be targets for cancer treatment. Trichostatin A [26] and ellagic acid [27], for example, increase CDKN1A expression and promote cell cycle arrest in bladder cancer cell lines; GADD45A expression was associated with the efficacy of the treatment with 5-aza-CdR in different pancreatic cancer cell lines [28]. Moreover, inactivation of CHEK1 appears to contribute to the development of cervical cancer [29], while increased E2F4 levels following genotoxic stress result in the downregulation of many mitotic genes and promote a G0-like state [30]. The inactivation of BRCA1 can be an important therapeutic target in sporadic breast cancers [31].
Three of the modulated genes, CDKN1A, GADD45A and CCNE1, are highlighted because they presented a fold change >2 (p < 0.05) in both cell lines. Therefore, their expressions were not associated with the type of TP53 mutation. The upregulation of CDKN1A and GADD45A might be respectively, related to the cell cycle arrest and apoptosis, observed in our previous study using the same cell lines [16]. Similarly, a number of authors has reported the induction of CDKN1A after gemcitabine treatment in MCF7 and MDA-MB231 cell lines (both from human breast carcinoma) [32]. Classically, the activation of TP53 gene induces CDKN1A mRNA increase, which leads to cell cycle arrest at the G1/S transition. However, it is important to emphasize that, even we have detected a TP53 overexpression in T24 cells after gemcitabine treatment, no significant alteration was observed in the 5637 cell line. T24 cells harbor TP53 mutations in the N-terminal transactivation domain which preserve p53 activities such as DNA binding. But in 5637 cells, TP53 has point mutations at core domain that affect the ability of p53 to bind DNA [33]. Thus, gemcitabine can induce an alternative mechanism of CDKN1A activation, disobeying the dogma that DNA damage activates TP53 gene and triggers CDKN1A accumulation, as Soria and Gottifredi [34] have already suggested for some genotoxic agents. With regard to the CCNE1 upregulation, it has been associated with a poor prognosis and resistance to chemotherapeutic drugs [35]. In fact, we have previously observed that TP53-mutated cells are more resistant to treatment with gemcitabine than the wild type [16]. Recently, Jiang et al. [36] explained that TP53 dysfunction compromises the nuclear export of wild-type BRCA1, thus characterizing the mechanism to increase cellular resistance to DNA damage in sporadic breast cancer. As far as we know, there are no data in literature about the modulation of the CCNE1 after gemcitabine treatment.
On the contrary, differential gene expression (with fold change >2) was also detected between the two cell lines. The BIRC5, CDK6, CUL2, HERC5, KPNA2, MAD2L1, NBN, RAD51, RB1, and TFDP2 genes were significantly modulated in the 5637 cells, while CCNB1, CCNF and TP53 were significantly modulated only in the T24 cells. Although most of these genes are associated with cell cycle regulation [37–39], their modulation was not solely responsible for the cell cycle arrest detected in the NDI experiments, since it occurred in both cell lines. Thus, the divergence between the gene expression signature in 5637 and T24 cell lines probably reflects the complexity of TP53 activity for regulating apoptosis and cell proliferation. Obviously, we cannot also rule out the possibility that other genes might be related to the observed differences in cell responses. In fact, TP53 is only one component of the giant surveillance network whose efficiency is modulated by many other elements, including other signaling pathways [40]. In a recent study using gene networks, we have described interactions among TP53 and several other genes, suggesting that other pathways might be associated with the malignant phenotype and chemotherapeutical response [41]. Furthermore, the results of this present study have showed different gene expression patterns after gemcitabine treatment, what suggest that combined evaluation of several genes may be required to finally predict gemcitabine sensitivity. We must remind, however, that not only differences in the gene signature were found in the present study. Some genes were similar and could explain the efficacy of treatment, independently of the genetic background of the cell lines analyzed. Therefore, TP53 status may not be related to some cell response to gemcitabine treatment.
To T24 cells, regarding the downregulation of the cyclin B1 (CCNB1) and cyclin F (CCNF) genes, they could be associated with a positive response to chemotherapy, as was observed in our study. Several authors have related the upregulation of these genes to the recurrence of the tumor [42, 43]. The increased risk for a sporadic benign meningioma recurrence has already been identified in cases with elevated expression of CCNB1 [42]. Furthermore, CCNF upregulation is characterized by reduced apoptosis and a more aggressive growth phenotype in HCT116 colorectal cancer cells that are resistant to the chemotherapeutic drug 5-fluorouracil [43].
Several genes were significantly upregulated in only the 5637 cells, including those related to cyclin-dependent kinases (CDK 6 and 8) and apoptosis inhibition (BIRC5). The upregulation of CDK6, CDK8 and BIRC5 have already been associated with cancer [44, 45]. However, it is important to notice that the balance between cell proliferation and cell death involves several mechanisms and that, sometimes, the signal for apoptosis surpasses the cell cycle signals. In fact, RB1, a tumor suppressor gene, and CDKN2B, which is involved in the negative regulation of the cell cycle, were also upregulated.
In conclusion, independent of the site of the TP53 mutation or the tumor grade, gemcitabine was cytostatic and modulated several molecular pathways, including DNA repair, G1/S transition, apoptosis and transcription factors. The identification of gemcitabine-responsive genes provides insight into its anti-cancer mechanisms and can be used in the clinical setting to predict chemotherapeutical responses. Further studies would be necessary to confirm whether the genes identified in the present study are associated with other types of cell growth inhibition after gemcitabine treatment.
References
Cordon-Cardo C (2008) Molecular alterations associated with bladder cancer initiation and progression. Scand J Urol Nephrol 218:154–165
Fajkovic H, Halpern JA, Cha EK, Bahadori A, Chromecki TF, Karakiewicz PI, Breinl E, Merseburger AS, Shariat SF (2011) Impact of gender on bladder cancer incidence, staging, and prognosis. World J Urol 29:457–463
Cheng L, Zhang S, MacLennan GT, Williamson SR, Lopez-Beltrn A, Montironi R (2011) FRCPath and IFCAP. Bladder cancer: translating molecular genetic insights into clinical practice. Human Pathol 42:455–481
Sylvester RJ, van der Meijden APM, Oosterlinck W, Witjes JA, Bouffioux C, Denis L, Newling DWW (2006) Predicting recurrence and progression in individual patients with stage TaT1 bladder cancer using EORTC risk tables: a combined analysis of 2596 patients from seven EORTC trials. Eur Urol 49:466–477
Nishiyama H, Watanabe J, Ogawa O (2008) p53 and chemosensitivity in bladder cancer. Int J Clin Oncol 13:282–286
Fujita H, Ohuchida K, Mizumoto K, Itaba S, Ito T, Nakata K, Yu J, Kayashima T, Souzaki R, Tajiri T, Manabe T, Ohtsuka T, Tanaka M (2010) Gene expression levels as predictive markers of outcome in pancreatic cancer after gemcitabine-based adjuvant chemotherapy. Neoplasia 12:807–817
Castagneto B, Zai S, Marenco D, Bertetto O, Repetto L, Scaltriti L, Mencoboni M, Ferraris V, Botta M (2004) Single-agent gemcitabine in previously untreated elderly patients with advanced bladder carcinoma: response to treatment and correlation with the comprehensive geriatric assessment. Oncology 67:27–32
Bellmut J, Albiol S, de Olano AR, Pujadas J, Maroto P (2006) On behalf the Spanish Oncology Genitourinary Group (SOGUG). Gemcitabine in the treatment of advanced transitional cell carcinoma of the urothelium. Ann Oncol 17:113–117
Shelley MD, Cleves A, Wilt TJ, Mason MD (2011) Gemcitabine chemotherapy for the treatment of metastatic bladder carcinoma. BJU Int 108:168–179
Toschi L, Finocchiaro G, Gioia V (2005) Role of gemcitabine in cancer therapy? Future Oncol 1:7–17
Ruiz Van Haperen VWT, Veerman G, Vermoken JB, Peters GJ (1993) 2′,2′-Difluoro-deoxycytidine (gemcitabine) incorporation into RNA and DNA from tumor cell lines. Biochem Pharmacol 46:762–766
Gontero P, Frea B (2006) Actual experience and future development of gemcitabine in superficial bladder cancer. Ann Oncol 17:123–128
Achanta G, Pelicano H, Feng L, Plunkett W (2001) Interaction of p53 and DNA-PK in response to nucleoside analogues: potential role as a sensor complex for DNA damage. Cancer Res 61:8723–8729
Bergman AM, Pinedo HM, Peters GJ (2002) Determinants of resistance to 2′,2′- difluorodeoxycytidine (gemcitabine). Drug Resist Updat 5:19–33
Galmarini CM, Clarke ML, Falette N, Puisieux A, Mackey JR, Dumontet C (2002) Expression of a non-functional p53 affects the sensitivity of cancer cells to gemcitabine. Int J Cancer 97:439–445
da Silva GN, Marcondes JPC, Camargo EA, Sakamoto-Hojo ET, Passos GA, Salvadori DMF (2010) Cell cycle arrest and apoptosis in TP53 subtypes of bladder carcinoma cell lines treated with cisplatin and gemcitabine. Exp Med Biol 235:814–824
Toshimitsu H, Iizuka N, Yamamoto K, Kawauchi S, Oga A, Furuya T, Oka M, Sasaki K (2006) Molecular features linked to the growth-inhibitory effects of gemcitabine on human pancreatic cancer cells. Oncol Rep 16:1285–1291
Pourquier P, Gioffre C, Kohlhagen G, Urasaki Y, Goldwasser F, Hertel LW (2002) Gemcitabine (20,20-difluoro-20-deoxycytidine), an antimetabolite that poisons topoisomerase I. Clin Cancer Res 8:2499–2504
Pearce HL, Miller MA (2005) The evolution of cancer research and drug discovery at Lilly, Research Laboratories. Adv Enzyme Regul 45:229–255
Coppée J-Y (2008) Do DNA microarrays have their future behind them? Microbes Infect 10:1067–1071
Fenech M (2007) Cytokinesis-block micronucleus cytome assay. Nat Protoc 2:1084–1104
Fencher G, Perabo FGE, Schmidt DH, Haase L, Ludwig E, Schueller H, Blatter J, Muller C, Albers P (2003) Preclinical evaluation of a radiosensitizing effect of gemcitabine in p53 mutant and p53 wild type bladder cancer cells. Urology 61:468–473
Aydemir N, Celikler S, Bilaloglu R (2005) In vitro genotoxic effect of the anticancer drug gemcitabine in human lymphocytes. Mutat Res 582:35–41
Jiang HT, Hickey RJ, Abdel-Aziz W, Malkas LH (2000) Effects of gemcitabine and araC on in vitro DNA synthesis mediated by the human breast cell DNA synthesome. Cancer Chemother Pharmacol 45:320–328
Attene-Ramos MS, Nava GM, Muellner MG, Wagner ED, Plewa MJ, Gaskins HR (2010) DNA damage and toxicogenomic analyses of Hydrogen Sulfide in human intestinal epithelial FHs 74 Int cells. Environ Mol Mutagen 51:304–314
Li G-C, Zhang X, Pan T-J, Chen Z, Ye Z-Q (2006) Histone deacetylase inhibitor trichostatin A inhibits the growth of bladder cancer cells through induction of p21waf1 and G1 cell cycle arrest. Int J Urol 13:581–586
Li T-M, Chen G-W, Su C-C, Lin J-G, Yeh C-C, Cheng K-C, Chung J-G (2005) Ellagic Acid Induced p53/p21 Expression, G1 Arrest and Apoptosis in Human Bladder Cancer T24 Cells. Anticancer Res 25:971–980
Missiaglia E, Donadelli M, Palmieri M, Crnogorac-Jurcevic T, Scarpa A, Lemoine NR (2005) Growth delay of human pancreatic cancer cells by methylase inhibitor 5-aza-2′-deoxycytidine treatment is associated with activation of the interferon signalling pathway. Oncogene 24:199–211
Mazumder D, Mitra S, Singh RK, Dutta S, Roy A, Mondal RK, Basu PS, Roychoudhury S, Panda CK (2011) Inactivation of CHEK1 and EI24 is associated with the development of invasive cervical carcinoma: clinical and prognostic implications. Int J Cancer 129:1859–1871
Pleska D, Crosby ME, Gupta D, Almasan A (2007) E2F4 function in G2: maintaining G2-arrest to prevent mitotic entry with damaged DNA. Cell Cycle 6:1147–1152
Galizia E, Giorgetti G, Piccinini G, Santinelli A, Loretelli C, Bianchi F, Gagliardini D, Carbonari G, Pisa E, Belvederesi L, Bracci R, Ferretti C, Corradini F (2010) BRCA1 expression in triple negative sporadic breast cancers. Anal Quant Cytol Histol 32:24–29
Hernandes-Vargas HH, Rodrigues-Pinilla SM, Julian-Tendero M, Sanchez-Rovira P, Cuevas C, Anton A, Rios MJ, Palacios J, Moreno Bueno G (2007) Gene expression profiling of breast cancer cells in response to gemcitabine: nK-kB pathway activation as a potential mechanism of resistance. Breast Cancer Res Treat 102:157–172
Cordon-Cardo C, Dalbagni G, Saez GT, Oliva MR, Zhang ZF, Rosai J, Reuter VE, Pellicer A (1994) p53 mutations in human bladder cancer: genotypic versus phenotypic patterns. Int J Cancer 56:347–353
Soria G, Gottifredi V (2010) PCNA-coupled p21 degradation after DNA damage: the exception that confirms the rule? DNA Repair 9:358–364
Lockwood WW, Stack D, Morris T, Grehan D, O’Keane C, Stewart GL, Cumiskey J, Lam WL, Squire JA, Thomas DM, O’Sullivan MJ (2011) Cyclin E1 is amplified and overexpressed in osteosarcoma. J Mol Diagn 13:289–296
Jiang J, Yang ES, Jian G (2011) Susceptibility to DNA damage p53-dependent BRCA1 nuclear export controls cellular. Cancer Res 71:5546–5557
Beukelaers P, Vandenbosch R, Caron N, Nguyen L, Belachew S, Moonen G, Kiyokawa H, Barbacid M, Santamaria D, Malgrange B (2011) Cdk6-dependent regulation of G(1) length controls adult neurogenesis. Stem Cells 29:713–724
Liu J, Vasudevan S, Kipreos ET (2004) CUL-2 and ZYG-11 promote meiotic anaphase II and the proper placement of the anterior-posterior axis in C. elegans. Development 131:3513–3525
Redwood AB, Gonzalez-Suarez I, Gonzalo S (2011) Regulating the levels of key factors in cell cycle and DNA repair: new pathways revealed by lamins. Cell Cycle 10:3652–3657
Soussi T, Wiman KG (2007) Shaping genetic alterations in human cancer: the p53 mutation paradigm. Cancer Cell 12:303–312
da Silva GN, Evangelista AF, Magalhães DA, Macedo C, Búfalo MC, Sakamoto-Hojo ET, Passos GA, Salvadori DM (2011) Expression of genes related to apoptosis, cell cycle and signaling pathways are independent of TP53 status in urinary bladder cancer cells. Mol Biol Rep 38:4159–4170
Bie L, Zhao G, Ju Y, Zhang B (2011) Integrative genomic analysis identifies CCNB1 and CDC2 as candidate genes associated with meningioma recurrence. Cancer Genet 204:536–540
de Angelis PM, Fjell B, Kravik KL, Haug T, Tunheim SH, Reichelt W, Beigi M, Clausen OP, Galteland E, Stokke T (2004) Molecular characterizations of derivatives of HCT116 colorectal cancer cells that are resistant to the chemotherapeutic agent 5-fluorouracil. Int J Oncol 24:1279–1288
Di Stefano AB, Iovino F, Lombardo Y, Eterno V, Höger T, Dieli F, Stassi G, Todaro M (2010) Survivin is regulated by interleukin-4 in colon cancer stem cells. J Cell Physiol 225:261–555
Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL (2011) Cyclin D as a therapeutic target in cancer. Nat Rev Cancer 11:558–572
Acknowledgments
This study was supported by FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo) and CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico), Brazil.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Glenda Nicioli da Silva and Elaine Aparecida de Camargo contributed equally to this study.
Rights and permissions
About this article
Cite this article
da Silva, G.N., de Camargo, E.A. & Salvadori, D.M.F. Toxicogenomic activity of gemcitabine in two TP53-mutated bladder cancer cell lines: special focus on cell cycle-related genes. Mol Biol Rep 39, 10373–10382 (2012). https://doi.org/10.1007/s11033-012-1916-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-012-1916-1