
Abstract
3′ addition events in miRNAs are widely detected and may contribute to miRNA stability, but little is known about details of the events in miRNA gene clusters and families. Here, we performed a comprehensive analysis of isomiR expression patterns and 3′ additions in miRNA gene clusters and families by analyzing high-throughput sequencing dataset. According to dominant modified isomiRs, miRNA members in many miRNA gene clusters and families showed the same 3′ additional non-template nucleotides. Although clustered miRNAs and homologous miRNAs had consistent or inconsistent expression levels, we found many of them showed consistent expression patterns at isomiR levels. These findings revealed similar processing mechanism and 3′ modification event of miRNAs in gene clusters and families through miRNA maturation process. The consistent maturation mechanism may contribute to co-regulate biological processes, and may originate from ancestral miRNA genes through complex duplication history.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
MicroRNAs (miRNAs), a class of prevalent small regulatory RNAs (~22 nt), mediate gene silencing and play key roles in various biological processes. Deregulation of specific miRNAs plays an important role in human diseases by regulating gene expression on a post-transcriptional level, including diverse cancers, and therefore miRNA profiling is proving invaluable in the study of human cancers. According to canonical pathway, miRNA is transcribed as a primary miRNA (pri-miRNA). The long transcript is then processed by Drosha and Dicer to produce the mature single-stranded miRNA, which is then loaded into the RNA-induced silencing complex (RISC) [1]. Due to the cleavage of Drosha and Dicer is imprecise and alternative, multiple miRNA variants with various 5′ and/or 3′ ends, termed as isomiRs, are generated from a given miRNA locus [2–5]. Interestingly, miRNAs may have additional non-template nucleotides at 3′ ends after Dicer processing [6–12]. The widespread phenomenon of 3′ additions is not a random event and conserved across animal species, especially prone to adenosines or uridines, which may contribute to miRNA stability and play a role in interactions of miRNA:target [7, 9]. However, isomiR profiles and 3′ addition events across different miRNAs in miRNA gene clusters and families still remain largely unknown.
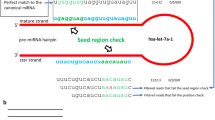
Some miRNA genes are linked as a cluster and as a single polycistronic transcript to coordinately regulate multiple biological processes. Clustered miRNA genes were estimated according to pairwise chromosomal distance at 10 kb, and these miRNA gene clusters were obtained according to annotation in the miRBase database. Some miRNA members in gene cluster share sequence similarity [13], and these miRNA members might experience complex duplication history [14–16]. With high sequence similarity, even with the same “seed sequence” (nucleotide 2–8), different miRNAs were named homologous miRNA genes and constituted gene family. Although miRNA cluster is often located in a polycistron and co-transcribed with neighboring miRNAs and host genes [17], they show consistent and inconsistent expression patterns [18, 19]. Here, by analyzing isomiR profiles and isomiR expression patterns of clustered miRNAs and homologous miRNAs, we address whether these special miRNAs have consistent maturation mechanism according to isomiR profiles and 3′ addition events.
Materials and methods
A human placental sample from pregnant women was obtained from Zhongda Hospital, Nanjing, China. Total RNA was extracted with TRIzol (Invitrogen), and small RNAs were isolated using mirVana® miRNA Isolation Kit (Ambion). Purified small RNAs were then subjected to miRNA library construction according to the protocol of SOLiD® Small RNA Expression Kit (ABI, Life Technologies). Sequencing was performed using SOLiD® system (ABI, Life Technology) at the State Key Laboratory of Bioelectronics, School of Biological Science and Medical Engineering, Southeast University, China. After discarding adaptor sequences, short RNAs were aligned to known human miRNA precursors (pre-miRNAs) sequences from the miRBase database (Release 16.0, http://www.mirbase.org/) [20] using Bowtie 0.12.7 [21]. 3′ non-template additional nucleotides were surveyed and compared based on miRNA members in gene clusters and families [5]. All of miRNAs were analyzed if their sequence counts were not less than 10. Relative expression levels of miRNAs were estimated based on sequence count of the most abundant isomiR and sum of all isomiR sequence counts, respectively. Due to the length distribution of miRNAs (~22 nt), shorter reads from deep sequencing can be mapped to multiple loci, termed as the phenomenon of cross-mapping in miRNAs, especially between multicopy miRNA precursors and homologous miRNA genes [16, 22] (Fig. S1). In order to comprehensively estimate relative expression levels of miRNAs in miRNA gene cluster and family, here counts of these common sequencing reads were repeatedly distributed between different miRNA genes. For example, hsa-miR-24 can be generated by hsa-mir-24-1 and hsa-mir-24-2, while the two mutlicopy miRNA precursors are found in different miRNA gene clusters (mir-23b gene cluster: mir-23b and mir-24-1; mir-24-2 gene cluster: mir-23a, mir-27a and mir-24-2). We hypothesized both the two pre-miRNAs could yield mature miRNAs, and therefore expression patterns of miRNAs in gene clusters could be estimated. In miRNA gene family, those miRNAs that can be yielded by multicopy pre-miRNAs only were estimated one time. According to several dominant isomiRs that can be generated from a given miRNA locus [5], we assessed whether miRNA expression patterns in gene clusters and families were consistent or not at isoimR levels by Fisher’s exact test. Specifically, the frequencies of high/low expressed isomiRs were estimated based on relative expression levels from a given miRNA locus. Rare isomiRs (less than 1% relative expression level) were removed from Fisher’s exact test.
Results
According to abundantly expressed miRNA clusters, we found most of their members always showed consistent expression patterns at isomiR levels based on Fisher’s exact test: one kind of dominant and abundant isomiR and other rare isomiRs, or several kinds of abundant isomiRs and other rare isomiRs (Fig. 1; Table S1). The fold change values of the most and secondary abundant isomiRs of clustered miRNAs always were similar (Table S2). According to abundant isomiRs, dominant cleavage sites of Drosha and Dicer during pre-miRNA processing could be estimated. These abundantly expressed isomiRs were not detected 3′ non-template additional nucleotides. Simultaneously, according to the dominant modified isomiRs, we found the same additional nucleotides between many clustered miRNAs (Table 1). Some clustered miRNAs might have different dominant additional nucleotides, but isomiR with consistent additional nucleotide always was secondary abundantly expressed isomiR. For example, hsa-miR-23b and hsa-miR-24-1 showed inconsistent dominant isomiRs with 3′ additions. However, expression level of hsa-miR-23b with adenosine was close to dominant isomiR with cytosine, which is consistent with dominant modified hsa-miR-24-1. Generally, isomiRs with 3′ additions had lower relative expression levels, and they showed larger expression divergence than dominant isomiRs from given miRNA loci. Interestingly, except for hsa-mir-143 cluster, others generated their mature miRNAs from the same arms of miRNA precursors. Based on different estimation methods, the most abundant isomiR and sum of all isomiRs, consistent and inconsistent expression levels of miRNA members in gene clusters could be found (Table 1). Specifically, some specific miRNAs had lower sequence counts, although their co-transcribed miRNAs in gene cluster were abundantly expressed (for example, hsa-mir-425 and hsa-mir-23b clusters, Table 1).


Consistent isomiR expression patterns between different miRNAs in gene clusters based on sequence counts. The horizontal coordinate every isomiR from the given miRNA locus. These isomiRs are ranked according to their relative expression levels. Except for several kinds of abundantly expressed isomiRs without non-template additional nucleotides, others always show rare sequence counts. Although inconsistent expression levels of miRNAs in gene cluster can be detected, they show consistent expression patterns according to their variants. Some members, with lower expression levels (less than 100), are not showed here
Similarly, in miRNA gene families, except for hsa-mir-103 family, other homologous miRNAs also showed consistent isomiR profiles and isomiR expression patterns (Fig. 2; Tables S1, S3). These homologous miRNAs were generated from the same arms of their miRNA precursors. Generally, at a given miRNA locus, isomiRs with 3′ additional nucleotides were not abundant isomiR. Many of them also showed dominant isomiRs with the same 3′ additional nucleotides (Table 2). Although homologous miRNA sequences always showed high sequence similarity, consistent and inconsistent expression patterns could be found. For example, hsa-miR-103 and hsa-miR-107 showed larger expression divergence (Table 2). On the other hand, we also estimated the dominant additional nucleotide based on sequence counts of all modified isomiRs from a single miRNA locus. As expected, clustered and homologous miRNAs always showed consistent dominant isomiRs with the same additional nucleotides (Tables S2, Table S3).

Consistent isomiR expression patterns between different miRNAs in gene families based on sequence counts. The horizontal coordinate every isomiR from the given miRNA locus. Except for several kinds of abundantly expressed isomiRs without non-template additional nucleotides, others always show rare sequence counts. Although inconsistent expression levels of miRNAs in gene cluster can be detected, they always show consistent expression patterns according to their variants (except for hsa-miR-103 and hsa-miR-107 in hsa-mir-103 family). Some members, with lower expression levels (less than 100), are not showed here
Discussion
Although consistent and inconsistent expression levels of miRNA members were detected in miRNA gene clusters and families (Tables 1, 2), they always showed consistent expression patterns at isomiR levels (Figs. 1, 2, Tables S1, S2, S3). Generally, 1–3 abundant isomiRs were detected from a given miRNA locus, but none of them was modified isomiR with 3′ non-template additional nucleotide [5]. Most of clustered miRNAs and homologous miRNAs showed consistent types of expressed abundant isomiRs and rare isomiRs (Figs. 1, 2; Table S1). Abundantly expressed isomiRs could be used to estimate dominant cleavage sites of Drosha and Dicer during pre-miRNA processing [23]. Consistent expression patterns implicated similar dominant cleavage mechanisms between these clustered miRNAs and homologous miRNAs. Also, these special miRNAs always were generated from the same arms of their pre-miRNAs. Due to modified isomiRs were not abundant isomiRs [5], 3′ non-template additional nucleotide was surveyed and compared based on dominant modified isomiRs. We found most of clustered miRNAs and homologous miRNAs showed dominant modified isomiRs with the same additional nucleotides, especially the miRNAs in gene clusters (Tables 1, 2). Adenosines and cytosines were dominant additional nucleotides in the study, and the phenomenon of 3′ addition event should not be a random event (Tables 1, 2). Some miRNA members showed inconsistent dominant modified isomiRs, for example, miRNAs in hsa-mir-143 cluster and hsa-mir-23 family. Interestingly, the additional nucleotide of one member could be detected as additional nucleotide of secondary abundantly expressed isomiR. Indeed, sometimes the relative expression levels of the most and secondary dominant modified isomiRs were very similar. Therefore, miRNA members in gene clusters and families had consistent additional nucleotides. Although these modified isomiRs only possessed lower percentage of total expression level from a given miRNA locus, some of them also unexpectedly showed higher sequence counts and relative expression levels. Simultaneously, we also estimated the most dominant additional non-template nucleotide according to sequence counts of modified isomiRs. The same nucleotide was detected as dominant additional nucleotide between miRNA members in many miRNA gene clusters and families (Tables S2, S3). 3′ addition events in miRNAs may play a role in miRNA stability and interactions of miRNA:target [7, 9]. The same dominant additional nucleotide in clustered miRNAs and homologous miRNAs may contribute to complex co-regulation network. Taken together, the interesting consistent isomiR expression patterns and dominant additional nucleotides might imply strict regulation during miRNA biogenesis in special miRNA groups (miRNA gene clusters and families).
In addition, we showed that the phenomenon of cross-mapping also contribute partly to the consistent isomiR profiles and expression patterns, especially between homologous miRNAs. Some clustered miRNAs were homologous miRNAs, for example, hsa-mir-30b cluster (including hsa-mir-30b and hsa-mir-30d genes). Due to high sequence similarity and various isomiRs with 5′ and/or 3′ ends, many isomiRs could be mapped to different miRNA genes (Fig. S1). Here, in order to perform a comprehensive analysis based on miRNAs members in gene clusters and families, these common isomiRs were repetitively estimated. Simultaneously, SOLiD sequencing platform employs a two-base encoding technology, and here only one mismatch was tolerated during mapping analysis using Bowtie software. To some extent, the strict selection of error can avoid cross-mapping events, but it still led to cross-mapping between homologous miRNA genes if they had common isomiR sequences, different nucleotides were located at 3′ ends or involved additional nucleotides (Fig. S1).
Despite of contribution of the cross-mapping, the fact that the interesting consistent isomiR expression patterns between miRNA members in gene clusters and families, especially consistent additional nucleotides of dominant modified isomiRs, indicated similar processing and maturation process and implicated specific evolutionary and functional relationships. Due to relative stability of 3′ addition events and isomiR profiles [5, 7], the consistent isomiR expression patterns and 3′ addition events in miRNA gene clusters and families were also supported by analyzing sequencing datasets of other placental samples. Therefore, the phenomenon should not be a random event. Clustered miRNA genes and homologous miRNA genes experience complex duplication history [14–16], and consistency of pre-miRNA processing and 3′ modification events, might be derived from their ancestral miRNA genes. The conservation of miRNA maturation mechanism would lead to similar isomiR expression patterns, including similar dominant modified isomiRs, which may contribute to co-regulate process. Further studies should reveal whether isomiR profiles and their expression patterns may contribute partly to the process of binding their targets, and consistent isomiR profiles, including widespread 3′ addition events, may also contribute to co-regulate biological process.
References
Kim VN, Han J, Siomi MC (2009) Biogenesis of small RNAs in animals. Natl Rev Mol Cell Biol 10:126–139
Kuchenbauer F, Morin RD, Argiropoulos B, Petriv OI, Griffith M et al (2008) In-depth characterization of the microRNA transcriptome in a leukemia progression model. Genome Res 18:1787–1797
Morin RD, O’Connor MD, Griffith M, Kuchenbauer F, Delaney A et al (2008) Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res 18:610–621
Ruby JG, Jan C, Player C, Axtell MJ, Lee W et al (2006) Large-scale sequencing reveals 21U-RNAs and additional microRNAs and endogenous siRNAs in C-elegans. Cell 127:1193–1207
Guo L, Yang Q, Lu JF, Li HL, Ge QY et al (2011) A comprehensive survey of miRNA repertoire and 3′ addition events in the placentas of patients with pre-eclampsia from high-throughput sequencing. PLoS One 6:e21072
Azuma-Mukai A, Oguri H, Mituyama T, Qian ZR, Asai K et al (2008) Characterization of endogenous human Argonautes and their miRNA partners in RNA silencing. Proc Natl Acad Sci USA 105:7964–7969
Burroughs AM, Ando Y, de Hoon MJ, Tomaru Y, Nishibu T et al (2010) A comprehensive survey of 3′ animal miRNA modification events and a possible role for 3′ adenylation in modulating miRNA targeting effectiveness. Genome Res 20:1398–1410
Ebhardt HA, Tsang HH, Dai DC, Liu YF, Bostan B et al (2009) Meta-analysis of small RNA-sequencing errors reveals ubiquitous post-transcriptional RNA modifications. Nucleic Acids Res 37:2461–2470
Fernandez-Valverde SL, Taft RJ, Mattick JS (2010) Dynamic isomiR regulation in Drosophila development. RNA 16:1881–1888
Lee LW, Zhang S, Etheridge A, Ma L, Martin D et al (2010) Complexity of the microRNA repertoire revealed by next generation sequencing. RNA 16:2170–2180
Li JJ, Yang ZY, Yu B, Liu J, Chen XM (2005) Methylation protects miRNAs and siRNAs from a 3′-end uridylation activity in Arabildopsis. Curr Biol 15:1501–1507
Katoh T, Sakaguchi Y, Miyauchi K, Suzuki T, Kashiwabara S et al (2009) Selective stabilization of mammalian microRNAs by 3′ adenylation mediated by the cytoplasmic poly(A) polymerase GLD-2. Genes Dev 23:433–438
Aravin AA, Lagos-Quintana M, Yalcin A, Zavolan M, Marks D et al (2003) The small RNA profile during Drosophila melanogaster development. Dev Cell 5:337–350
Hertel J, Lindemeyer M, Missal K, Fried C, Tanzer A et al (2006) The expansion of the metazoan microRNA repertoire. BMC Genomics 7:25
Zhang YF, Zhang R, Su B (2009) Diversity and evolution of MicroRNA gene clusters. SCI China C 52:261–266
Guo L, Lu Z (2010) Global expression analysis of miRNA gene cluster and family based on isomiRs from deep sequencing data. Comput Biol Chem 34:165–171
Baskerville S, Bartel DP (2005) Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA 11:241–247
Yu J, Wang F, Yang GH, Wang FL, Ma YN et al (2006) Human microRNA clusters: genomic organization and expression profile in leukemia cell lines. Biochem Biophys Res Commun 349:59–68
Viswanathan SR, Mermel CH, Lu J, Lu CW, Golub TR et al (2009) microRNA expression during trophectoderm specification. PLoS One 4:e6143
Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ (2008) miRBase: tools for microRNA genomics. Nucleic Acids Res 36:D154–D158
Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:212
de Hoon MJL, Taft RJ, Hashimoto T, Kanamori-Katayama M, Kawaji H et al (2010) Cross-mapping and the identification of editing sites in mature microRNAs in high-throughput sequencing libraries. Genome Res 20:257–264
Guo, L, H Li, J Lu, Q Yang, Q Ge et al (2011) Tracking miRNA precursor metabolic products and processing sites through completely analyzing high-throughput sequencing data. Mol Biol Rep 39:2031–2038
Acknowledgments
The work was supported by the project 30871393 from National Natural Science Foundation of China and funded by Tsinghua National Laboratory for Information Science and Technology (TNList) Cross-discipline Foundation. The work was also supported by a research Grant from the Innovation Project for Graduate Student of Jiangsu Province (No. CX10B_081Z), the Scientific Research Foundation of Graduate School of Southeast University, Science and Technology Project in Nanjing (201001095) and Pre-Research Project for National Natural Science Foundation supported by Southeast University (KJ2010442).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Li Guo, Hailing Li, and Tingming Liang contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
11033_2012_1493_MOESM1_ESM.tif
Fig. S1 The contribution of cross-mapping between homologous miRNAs. The three isomiRs can simultaneously map to hsa-miR-23a and hsa-miR-23b. (TIFF 34 kb)
Rights and permissions
About this article
Cite this article
Guo, L., Li, H., Liang, T. et al. Consistent isomiR expression patterns and 3′ addition events in miRNA gene clusters and families implicate functional and evolutionary relationships. Mol Biol Rep 39, 6699–6706 (2012). https://doi.org/10.1007/s11033-012-1493-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-012-1493-3