
Abstract
Six full-length cDNA encoding boron transporters (BOR) were isolated from Brassica napus (AACC) by rapid amplification of cDNA ends (RACE). The phylogenic analysis revealed that the six BORs were the orthologues of AtBOR1, which formed companying with the triplication and allotetra-ploidization process of B. napus, and were divided into three groups in B. napus. Each group was comprised of two members, one of which was originated from Brassica rapa (AA) and the other from Brassica oleracea (CC). Based on the phylogenetic relationships, the six genes were named as BnBOR1;1a, BnBOR1;1c, BnBOR1;2a, BnBOR1;2c, BnBOR1;3a and BnBOR1;3c, respectively. The deduced BnBOR1 s had extensive similarity with other plant BORs, with the identity of 74–96.8% in amino acid sequence. The BnBOR1;3a and BnBOR1;3c resembled AtBOR1 in number and positions of the 11 introns, but the others only have 9 introns. After the gene duplication, there was evidence of purifying selection under a divergent selective pressure. The expression patterns of the six BnBOR1 s were detected by semi-quantitative RT–PCR. The BnBOR1;3a and BnBOR1;3c showed a ubiquitous expression in all of the investigated tissues, whereas the other four genes showed similar tissue-specific expression profile. Unlike the non-transcriptional regulation of AtBOR1, the expression of BnBOR1;1c and BnBOR1;2a were obviously induced by boron deficiency. This study suggested that the BOR1 s had undergone a divergent expression pattern in the genome of B. napus after that the B. napus diverged from Arabidopsis thaliana.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Boron (B) is an essential microelement for higher plants [1], and the new evidences suggested that B is also essential or beneficial for several animals, including humans [2]. Most of B is localized in cell wall of plants, which plays an important function for the maintenance of cell wall integrity by its cross-linking with rhamnogalacturonan II (RG-II) in pectins [3–6]. The B requirement of plants is low but significantly different among various species. In general, monocots need less B for normal growth and development and show higher tolerance to B deficiency than dicots [7, 8].
In the early studies, it was widely believed that passive diffusion was a major or possibly only mechanism of B transmembrane transport, and B translocation in plants was also a passive transport by the transpiration stream [9–11]. However, the physiological experiments revealed that the channel-mediated diffusion and energy-dependent active transport were involved in B transport [12, 13]. Recently, several members of the major intrinsic proteins (MIPs) have been established as the boric acid channels in plants [14–18]. In addition, the first B transporter, AtBOR1, was identified for B xylem loading in Arabidopsis thaliana [19]. The AtBOR1 mediated B exporting from pericycle cells into the root stelar apoplast against a concentration gradient. Both of boric acid channel and boron transporter were shown to be required for plant growth, especially under B limitation [20]. The AtBOR1 was not affected at the transcriptional level by B supply but regulated by posttranscriptional mechanisms. Under B deficiency, the protein of AtBOR1 accumulated in plasma membrane, whereas under normal or high B supply, it was internalized through endocytosis and degraded in vacuole [21]. OsBOR1, which was similar to AtBOR1 in rice, participated in efficient B transport at low B environments [22]. Under high B supply, Bot1, an AtBOR1-like protein isolated from wheat and barley, alleviated B toxicity by excreting B out of root cells [23]. Positive correlations between mRNA levels and tolerance of high B showed that the Bot1 played an important role in tolerance to high B toxicity. The same phenomenon was observed in A. thaliana, which the overexpression of AtBOR4 improved B tolerance by excluding B out of the roots. [24]. There is a narrow range between B-deficient and B-excessive levels, and the molecular mechanisms of B transport by BORs maintain B homeostasis for plants growth and development. The further studies showed that AtBOR1 and AtBOR4 had a different location in plasma membrane. AtBOR1 is localized preferentially in the proximal side of cells, in contrast, AtBOR4 is localized in distal side in epidermal cells [20, 24]. The polarized localization of borate exporters suggested the directional transcellular transport of B in plants. The chimera of AtBOR1 and AtBOR4 revealed a sorting motif involved in AtBOR1 endocytic and polarized trafficking [25, 26].
Genes similar to AtBOR1 were found in wide range of eukaryotes but not in prokaryotes. The B transporter belongs to the bicarbonate transporter superfamily (SLC4), which was known as band3 (SLC4A1) and well researched in erythrocyte [19, 27]. In mammals, NaBC1 (or SLC4A11) was identified as a special electrogenic Na+-coupled borate transporter [28]. YNL275w in Saccharomyces cerevisiae, one homolog to AtBOR1, was also characterized to be efflux B transporter in plasma membrane [16]. Recently, another type of boron transporter, ATR1, has been found in S. cerevisiae, which encodes a multidrug resistance transport protein. ATR1 was upregulated by boron supply in transcriptional level and required for boron tolerance [29].
The cultivated Brassica species are most closely related to A. thaliana, all of which are members of the Brassiceae tribe within the Brassicaceae family. The analysis of the A. thaliana genome revealed that there were at least three ancient polyploidy events. The most recent event was called the At-α (alpha) or 3R, the intermediate event was referred to as the At-β (beta) or 2R, and the oldest was the At-γ (gamma) or 1R.The Brassica genomes shared the three round ancient polyploidization events with A. thaliana and had a genome-wide triplication events soon after divergence from A. thaliana about 15 million years ago (MYA) [30]. Brassica napus (2n = 38, AACC) is a young allotetraploid formed from the recent fusion of two diploid genomes, an A-genome progenitor (Brassica rapa, 2n = 20, AA) and a C-genome progenitor (Brassica oleracea, 2n = 18, CC) [31]. The diploid progenitors’ genome triplication and allotetra-ploidization resulted in the multiple copies of genes in the AACC genome and equal numbers of genes in B. napus occur as homeologous pairs, one from the A-genome and the other from C-genome [32].
Oilseed rape (B. napus L.), one of the most important oil crops in the world, have a high requirement for B. It usually requires more than 0.5 mg kg−1 of hot water-soluble B (HWSB) in soils to complete its growth and development [33]. Soils with low available B were widespread, and which became one of the limiting factors for the sustainable production and high quality of oilseed rape in many agricultural areas of the world. However, it has been found that there was significant difference among cultivars of B. napus in their response to B-deficiency [34].
In the present study, six orthologues of AtBOR1 were isolated and characterized from B. napus, and further their structure, expression pattern and the evolutionary history were analyzed.
Materials and methods
Plant materials and culture conditions
Seeds of Brassica napus cv. Qingyou10 (QY10) used in this study were germinated after surface-sterilized with 0.5% NaClO (w/v) solution for 15 min and washed in deionized water. The uniform seedling were cultivated in a greenhouse under 24°C 8/16 h light/dark cycles with a relative humidity about 65–80% for 3 weeks, then the plants were sampled for DNA and RNA isolation. On the other hand, QY10 was grown under field conditions with two B treatments and three replicates. The field soil is an acid soil with pH 4.8. It contained 4.6 g kg−1 organic matter, 98.01 mg kg−1 alkaline hydrolytic nitrogen, 2.85 mg kg−1 Olsen-P, 31.48 mg kg−1 NH4Cl-exchangeable K and 0.12 mg kg−1 hot water-soluble boron, which belongs to severe B-deficient level. The application amount of N, P, and K fertilizers was calculated according to the following nutrient rates: 120 kg ha−1 N (urea, N 46%), 90 kg ha−1 P2O5 (calcium superphosphate, P2O5 12%), 150 kg ha−1 K2O (potassium chloride, K2O 60%). For the normal B treatment, additional 15 kg ha−1 borax was applied. For the B deficiency treatment, additional 0.75 kg ha−1 borax was applied by spraying foliage at seedling stage. The roots, stems, leaves, buds, flowers and siliques were sampled at bloom stage for the investigation of genes expression. All samples were immediately frozen in liquid nitrogen, and stored at −80°C.
Nucleic acid isolation
Total genomic DNA was extracted from mixed fresh leaves about 5 g using a CTAB method [35]. Total RNA was extracted from different tissues using TRIzol reagent (Invitrogen, USA). Each RNA sample was treated with RNase-free DNase I (Worthington, USA) to eliminate contaminated DNA. Agarose gel electrophoresis and spectrophotometer analysis were used to determine the quality and quantity of nucleic acid samples. These RNA were then used as template for reverse transcription reaction with the M-MLV reverse transcriptase and oligo dT(18) primer. 2 μg of total RNA was reversely transcribed using 0.5 μg of oligo dT(18) primer, 0.5 mM dNTPs and 200 U M-MLV Reverse Transcriptase (Promega, USA) at 42°C for 1 h in the appropriate buffer. The reaction was stopped by incubation at 70°C for 10 min. 2 μl of the first strand cDNA was used as template for semi-quantitative RT–PCR with ExTaq (Takara, Japan).
Rapid amplification of 3′- and 5′-cDNA ends (RACE) of target gene
Based on the sequence of AtBOR1 and its paralogous genes, two degenerate primers, DeGF and DeGR, were designed (Table 1). The first production of the polymerase chain reactions (PCR) was screened and the positive clones were sequenced for obtaining a fragment homologous to AtBOR1. According to the sequence information, four gene specific primers, RACE3-1, RACE3-2, RACE5-1 and RACE5-2, were designed for 3′-RACE and 5′-RACE, respectively (Table 1). 2 μg of total RNA was used as template to generate first-strand cDNA in terms of the RACE user manual (GeneRacer kit, Clontech, USA). Two sense primers, RACE3-1 and RACE3-2, were paired with kit 3′-Primer and 3′-Nested Primer for primary and nested amplifications of 3′-cDNA ends, respectively. Similarly, two antisense primers, RACE5-1 and RACE5-2, for the 5′-cDNA ends amplifications. The PCR was conducted in 50 μl standard amplification system containing 2.0 units of KOD-Plus DNA Polymerase (Toyobo Co., LTD) and other ingredients. In primary amplification, 2 μl of total first-strand cDNA was used as template. Then, 2 μl of the primary amplification product was diluted 100 times and 2 μl of the diluted production was used for the nested amplification. The PCR was carried out on a MyCycler gradient thermocycler (BioRad, USA) under following conditions: predenaturation at 94°C for 3 min, followed 30 cycles: 94°C for 30 s, 70°C for 30 s, 72°C for 2 min, and succeeded by a final extension at 72°C for 10 min. The PCR products were electrophoresis-analyzed on 1% agarose gel in 1× TAE, ethidium bromide stained, and UV-visualized. DNA of target bands was recovered and ligated to the pGEM-T vector (Promega, USA). Escherichia coli strain DH5α was transformed by standard CaCl2 method and screened. The PCR-positive white colonies were sequenced.
Amplification of full-length cDNAs and corresponding genomic sequences
Based on the 5′- and 3′-cDNA ends obtained, sense primer BF1 and antisense primer BR1, BR2 and BR3 were synthesized to amplify the region containing the open reading frame (ORF). 2 μl of total cDNA as template and 1.0 units of KOD-Plus DNA Polymerase (Toyobo Co., LTD) were used in a 25 μl PCR reaction system. PCR cycling conditions were as follows: predenaturation at 94°C for 3 min, followed by 35 cycles: 94°C for 30 s, 68°C for 30 s, and 72°C for 3 min, and then succeeded by 10 min at 72°C. Genomic sequences were amplified using the same conditions by substituting the template with 0.1 μg of total genomic DNA. Target bands were recovered and subcloned, and the PCR-positive white colonies were subjected to further screening. The positive colonies were sequenced.
Database search and sequence alignments
Sequence blast analyses were conducted on NCBI web site (http://www.ncbi.nlm.nih.gov/). The obtained cDNA and genomic sequences BlastN against green plants nr database, respectively. The sequences were retrieved to local database according to the hits E-value below 10−10 and the identity score above 50%. The sequences of BnBOR1 s and the sequences retrieved were loaded into BioEdit and the local reciprocal blast was used for identify orthologues [36, 37], i.e. blasting the gene aa from AA species against BB species database and then blasting the gene bb from BB species against the AA species database. If both the results showed that two genes, aa and bb, had the highest hits, the two genes were presumed orthologues. Then, the coding sequences (CDS) of these sequences were translated into proteins and aligned using ClustalW [38]. Alignments were carefully checked, and the obviously alignment mistakes were corrected manually. Both the identity values of nucleotide and protein sequence from all the possible comparisons were obtained using the BioEdit. Protein structure predictions were carried out on Expasy web site (http://www.expasy.org).
Phylogenetic analysis
The phylogenetic trees were constructed by the Neighbor-Joining method with MEGA 4.0 [39]. The ambiguously aligned regions of putatively functional BOR genes were removed, leaving 85–90% coding regions for phylogenetic analyses. Before phylogenetic trees were constructed, all of the sequences were initially subjected to the chi-square analysis for homogeneity of base composition implemented in Tree-Puzzle (version 5.2) [40]. The reliability of the tree was measured by bootstrap analysis with 1,000 replicates.
RT–PCR detection of transcription levels
Semi-quantitative RT–PCR was carried out to detect the transcription levels of the BnBOR1 s members in various tissues of B. napus. 5 μl total first-strand cDNA of each sample was used as template in a 50 μl standard Taq PCR reaction system. The PCR were performed as following parameters: 94°C for 3 min; then 35 cycles at 94°C for 5 s, 50°C for 10 s, 72°C for 15 s; and finally with an extension at 72°C for 3 min. Primers used for the specific gene in RT–PCR were listed in Table 1. Quantitative and qualitative uniformity of the cDNA samples was monitored by a 24 cycle PCR using positive internal control with primers 18SF and 18SR (Table 1), which amplified 188 bp fragment from the B. napus 18S rRNA. 10 μl of the amplified PCR products were electrophoresed on a 1.2% (w/v) agarose gel with 0.5 μg ml−1 ethidium bromide in 1× TAE and UV-visualized using the FR-980 Bio-Electrophoresis Image Analysis System. All of the semi-quantitative RT-PCRs were carried out with three replicates.
Molecular evolutionary analysis
In order to estimate the form and strength of selection pressure on the BnBOR1 s gene family after gene duplication, we used maximum likelihood methods to estimate dN/dS (or ω), the ratio of the rates of nonsynonymous (dN) and synonymous (dS) nucleotide substitutions, by employing a variety of codon substitution models as implemented in the codeml program of the PAML 4.4 software package [41].
We fit three nested models to test for variation in selection pressure and/or positive selection [42]. First, Branch models, which allow the ω ratio to vary among branches in the phylogeny, were used to test for episodic adaptive evolution after gene duplications [43]. The following models were compared: M0 (one ω ratio for all branches) with the two-ratio model (TR, the foreground branches have a different ω ratio from the background branches). Second, to test the presence of the positive selection on individual codon sites, site models were applied that hold ω constant among all branches but allow ω to vary among codons [44, 45]. The following models were compared: M7 (beta, a model of beta-distributed selective pressures, which allows for 10 site classes, each with ω < 1) with M8 (beta and ω, a model with 11 site classes, one of which allows ω > 1). We identified the sites under the positive selection with the Bayesian approach under M8. Third, to test the presence of the different selective pressure between the genes, the following pairs of models were implemented. The nearly neutral model (M1a) assumes two classes of sites: one is under purifying selection with 0 < ω 0 < 1, the other is under neutral evolution with ω 1 = 1, and was compared to Clade Model C, which includes three site classes and five parameters. The ω variation among branches and sites was detected by employing a likelihood ratio test (LRT) between two nested models (null and alternative models). In the LRT, twice the log likelihood difference between the two models was compared with the chi-square statistics with degrees of freedom (df) equal to the difference of the number of parameters, i.e., 2 df both for M0/TR and M7/M8 tests. If the LRT was statistically significant with the ω ratio >1, it suggests that the positive selection was contributed to the evolution of genes below groups.
All PAML analyses were carried out using the F3 × 4 codon frequency estimation methods. The tree branch lengths were estimated under the M0 model and then used as the initial values in more complex codon model analyses. To prevent incorrect parameter estimates for local optima and low accuracy in Bayesian identification of the positively selected codon sites, the codeml program was run multiple times specifying different initial ω values (below, at, and above ω = 1) in order to check for convergence. The results of runs with the best likelihood scores were only considered further.
Results and discussion
Cloning of BnBOR1 genes in Brassica napus
Genes encoding BORs have been cloned and characterized from some higher plant species, such as A. thaliana, O. sativa, wheat (Triticum aestivum), barley (Hordeum vulgare) and eucalyptus trees because of its importance in the alleviation of both B deficiency and B toxicity. After total RNA was isolated from B. napus leaves and then reversely transcribed into cDNA, the degenerate primers (DeGF and DeGR in Table 1) were used to specifically amplify a 1042 bp product by RT-PCR. A blastn search showed that the PCR product was homologous to BOR genes from other plant species. Then, four gene-specific primers as described above were designed and synthesized for the 3′-RACE and 5′-RACE based on the 1042 bp fragment. Nested PCR of 3′-RACE produced a band of about 1700 bp. The sequences analysis showed that there were seven different 3′-cDNA ends, two of which showed alternative polyadenylation sites. Nested PCR product of 5′-RACE showed a band of about 800 bp, and two different 5′-cDNA ends were obtained. Orthology of these cDNA ends to AtBOR1 was proved by BlastN in NCBI. Based on the 5′- and 3′-cDNA ends, three primer pairs (BF1 versus BR1, BR2 and BR3) were designed to amplify the full-length of the BnBOR1 genes. With total cDNA as template, three specific bands about 2.2 kb were produced. Meanwhile, genomic sequence amplifications yielded specific bands about 3.0 kb. After sequenced, six unique full-length cDNA and corresponding genomic sequences were obtained and named as BnBOR1 s. The nucleotide sequences reported in this work have been submitted to the GenBank with the accession numbers GU827643.1 to Gu827648.1 for mRNA and GU827651.1 to GU827656.1 for genomic sequences, respectively, which were listed in Table 2.
Characterization of the six BnBOR1 paralogues
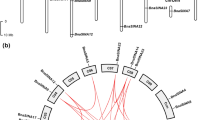
A nomenclature was proposed that the six genes were named as BnBOR1;1a, BnBOR1;1c, BnBOR1;2a, BnBOR1;2c, BnBOR1;3a and BnBOR1;3c, respectively, which was explained in describing the phylogenetic relationships below. The full-length cDNAs and genomic sequences of the six BnBOR1 s members were 2459–2769 and 2903–3147 bp, respectively, and the 5′-UTR length was ranged from 46 to 390 bp and its length of ORF (including stop codon TAA) was between 2106 and 2115 bp, and 3′-UTR 162–378 bp, respectively (Table 2). Start- and stop-codon positions of the BnBOR1 s genes were identical to those of AtBOR1, except for 3 bp premature stop codon of BnBOR1;3a and BnBOR1;3c. The two BnBOR1;3 s resembled AtBOR1 in number and positions of the 11 introns, but the other four genes have two introns loss (Fig. 1a). Therefore, they contained 9 introns, of which introns 1–4 and 5–9 corresponded to introns 2–5 and 7–11 of AtBOR1, respectively. All of the introns were followed by standard GT…AG splicing boundaries. Similar to AtBOR1, mature mRNAs of BnBOR1;2a and BnBOR1;2c encoded proteins of 704 amino acids (aa) (Table 2). Due to three amino acids gap in C-terminal region, the inferred BnBOR1;1a and BnBOR1;1c proteins had 701 amino acids. The BnBOR1;3a and BnBOR1;3c had 703 amino acids for 3-bp premature termination codon (Table 2). The BnBOR1 proteins possessed theoretical molecular weights (MWs) of 78.24–78.64 kDa and isoelectric points (pIs) of 8.68–8.98 (Table 2).

Identification and sequences analysis of BnBOR1s. a The schematic of the BnBOR1s structure. E extron, I intron, the broken line of I1 and I6 indicated one intron loss. b Hydrophobicity plots of BnBOR1;1a. Plots were generated as described by Kyte and Doolittle [46]. The running window was 13 amino acids conducted on BioEdit 7.0.9.0. The 10 putative transmembrane domains were marked with a black bar
Comparisons analysis among the BnBOR1 s showed high identity both at nucleotide and amino acid levels (Table 3). Based on sequence similarity and gene structure, BnBOR1 genes could be divided into three groups. The nucleotide identity between BnBOR1;1a and BnBOR1;1c in the group I is 96.9% and in the group II (BnBOR1;2a, BnBOR1;2c) is 96.4%. Group I and Group II showed 93.0–93.7% identity each other. In the group III, BnBOR1;3a was 97.7% identical to BnBOR1;3c. Group III shared 89.9–92.3% identity with Group I and Group II (Table 3). Comparison of the BnBOR1 s with AtBOR1 showed 89.1–91.3% identity on nucleotide level and 95.5–96.8% similarity on the amino acid level, respectively (Table 3). Protein–protein BlastP and multiple alignment analysis showed the deduced BnBOR1 s amino acid sequence had high similarity with BOR sequences from other plant species, such as AtBOR1 (>95% A. thaliana), OsBOR1 (86% O. sativa) and HvBOR2 (>83% H. vulgare), suggesting that BnBOR1 s belonged to the BORs family. Conclusively, BnBOR1 s proteins are typical plant BOR proteins with high similarities to AtBOR1.
The hydrophobicity profiles were generated with 13 amino acids running window using the general method of Kyte and Doolittle [46], and the result showed there were at least 10 trans-membrane domains along the BnBOR1 s (Fig. 1b). The BnBOR1 s contained a cotransporter motif, which was similar to other BORs reported previously [16, 19, 22–24, 28]. A pfam00955 (HCO3_cotransp) conserved domain, was detected to locate at M1–R (or H) 180, K220–D335 and V458–F549 of BnBOR1 s by NCBI conserved domain (CD) search (Supplementary material, Fig. S1). The secondary structure of BnBOR1 s was analyzed by SOPMA [47] and the result showed that the putative BnBOR1 s peptide contained 44.46% of alpha helices, 14.49% of extended strands, 3.55% of beta turns and 37.50% of random coils. Penetrating through most parts of the secondary structure, alpha helices and random coils were the most abundant structural elements in BnBOR1 s, while extended strands and beta turns were intermittently distributed in protein.
Phylogenetic tree and proposed nomenclature of BnBOR1 s
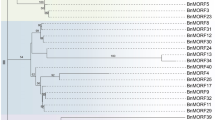
The phylogenetic relationships among BOR-like genes from B. napus, A. thaliana and O. sativa were analyzed with a phylogenetic tree constructed from the nucleotide sequences of these genes. The primary sequences were obtained from GenBank (accession numbers indicated in brackets): AtBOR1 [NM_180138.2], AtBOR2 [NM_116092.3], AtBOR3 [NM_001125118.1], AtBOR4 [NM_101415.3], AtBOR5 [NM_106139.2], AtBOR6 [NM_122453.4], AtBOR7 [NM_119403.5], OsBOR1 [NM_001073581.1], OsBOR2 [NM_001048710.1], OsBOR3 [NM_001048709.1] and OsBOR4 [DQ421409]. All of the BnBOR1 s members formed a small branch with a 100% bootstrap support (Fig. 2a), which were further clustered with AtBOR1 to form a distinct Brassicaceae BOR1 s branch. It was clear that BnBOR1 s were the orthologous genes to AtBOR1 in B. napus. Brassica BOR1 genes were separated into three distinct groups with 100% bootstrap support: BnBOR1;1a and BnBOR1;1c in Group I, BnBOR1;2a and BnBOR1;2c in Group II, and BnBOR1;3a and BnBOR1;3c in Group III. The group I is closely related to the group II. B. napus (AACC) is an allotetraploid formed from the recent fusion of two diploid genomes, an A-genome progenitor (B. rapa, AA) and a C-genome progenitor (B. oleracea, CC). In order to determine whether the gene was originated from AA genome or CC genome, the reciprocal blast was conducted in local database. The result indicated that the BnBOR1;1a, BnBOR1;2a and BnBOR1;3a were originated from the AA genome and the others from the CC genome. Another phylogenetic tree was conducted with BnBOR1 s and six sequences (DU112151.1, EX081325.1 and EX076122.1 are from B. rapa, and AM058271.1, DY025746.1 and AM057375.1 are from B. oleracea) (Fig. 2b), which were identified as the orthologous fragment by reciprocal blast and selected for overlapping on 1486-2106 of BnBOR1;1a coding region. The phylogenetic tree showed the same topology structure as the previous one with a lower bootstrap value support for a limited polymorphism (shorten length and high similarity).

Phylogenetic analysis of BnBOR1 s a Phylogenetic relationships between BnBOR1 s and other plants’ BORs: A. thaliana AtBOR1 [NM_180138.2], AtBOR2 [NM_116092.3], AtBOR3 [NM_001125118.1], AtBOR4 [NM_101415.3], AtBOR5 [NM_106139.2], AtBOR6 [NM_122453.4], AtBOR7 [NM_119403.5]; O. sativa OsBOR1 [NM_001073581.1], OsBOR2 [NM_001048710.1], OsBOR3 [NM_001048709.1] and OsBOR4 [DQ421409]. b Phylogenetic tree of BnBOR1 s and its orthologous fragment detected by reciprocal Blast in B. rapa and B. oleracea. Both phylogenetic trees were constructed by Neighbor-Joining method based on the codon nucleotide sequences. The number for each interior branch was the percent bootstraps value (1000 replicates). The scale bar indicated the estimated number of nucleotide substitutions per site
The six BnBOR1 s genes were the orthologues of AtBOR1 and divided into three groups based on the phylogenetic analysis (Fig. 2). The reciprocal blast suggested that each group was comprised of two members, one of which is from B. rapa and the other from B. oleracea. The result implied that these six genes were formed companying with the triplication and allotetra-ploidization of the B. napus. Combining the reciprocal blast and phylogenetic result, a nomenclature was proposed that the six genes were named BnBOR1;1a, BnBOR1;1c, BnBOR1;2a, BnBOR1;2c, BnBOR1;3a and BnBOR1;3c, respectively. Bn denoted B. napus, BOR1 denoted that the gene is an orthologues of AtBOR1, the number of 1, 2, 3 followed the semicolon denoted the three groups, and a or c denoted that the gene was originated from the B. rapa (AA) or B. oleracea (CC) genome.
Expression profile of BnBOR1 s in different tissues of B. napus
To investigate the expression pattern of BnBOR1 s, total RNA was isolated from different tissues including roots, stems, leaves, flowers, buds and siliques grown under normal and B-deficient conditions, respectively, and then it was subjected to semi-quantitative RT–PCR analysis. The intensity of the amplified products indicated variable levels of BnBOR1 s expression in different tissues. The genes from the same group displayed a similar tissue-specific expression profile (Fig. 3). The expression of Group III genes could be detected in all tested tissues, while that of both Group I and II genes showed similar tissue-specific expression profile, which expressed strongly in roots and stems, moderately in flowers, weakly in buds, and undetectable in leaves and siliques. The BnBOR1;1a mainly expressed in stems, moderately in roots and flowers under normal B condition, while the BnBOR1;1c expressed most abundantly in roots. In comparison with BnBOR1;1 s, BnBOR1;2 s showed the strongest expression in stems and a detectable expression in buds under normal B condition. In the case of B deficiency condition, the accumulation of BnBOR1;1c was up-regulated distinctly in roots, stems and flowers (Fig. 3). The expression of BnBOR1;2a, like BnBOR1;1c, was up-regulated in roots, stems and flowers. The highest expression of BnBOR1;3a was observed in roots, while the BnBOR1;3c was constitutively expressing in all tested tissues regardless of B supply (Fig. 3).

Semi-quantitative RT–PCR analysis of BnBOR1 s in different tissues under boron deficiency and normal conditions The different tissues Ro root, St stem, Le leaf, Fl flower, Bu bud, Si silique under boron deficiency (−B) and normal condition (normal)
In Arabidopsis, AtBOR1 was mainly expressed in the root pericycle cells and exports B from stele cell to xylem [19]. This was the key step of B translocation from root to shoot. Unlike the non-transcriptional regulation of AtBOR1, the expression of BnBOR1;1c and BnBOR1;2a were induced in the case of B deficiency (Fig. 3). This presented a major difference between the two BnBOR1 s and AtBOR1. Several reports showed that the nutrient transporters were induced by the nutrient deprivation, such as IRT1 in rice [48] and YSL2 in Arabidopsis [49]. The expression of the BnBOR1;3a and BnBOR1;3c could be detected in all tested tissues which indicated that the group III genes had ubiquitous expression profile (Fig. 3). The spatial expression patterns of BnBOR1;3 s implied that Group III genes played a basal role in B translocation in B. napus. Moreover, it suggested that a general function of B transport was not only for B xylem loading by roots as AtBOR1 in Arabidopsis [19, 22], but also for a more widespread translocation of B into different cell types in multiple tissues, which includes parenchymal and vascular tissues. All of the six genes were expressed in roots, stems and flowers, which reflected the three B translocation process in these tissues. The root was involved in B uptake and xylem loading, the stem in B long distance transport and the flower in pollen tube elongation, which would be influenced by the B supply. The leaf was also involved in B translocation in previous reports. In this study, besides the ubiquitous expression of the group III genes, the others were an undetectable level in leaves (Fig. 3). This may be explained that B was transported through transpiration stream by MIPs and BORs play a less important role of B translocation in leaves. The BnBOR1;3a displayed the highest expression in roots which implied that it was mainly involved in B uptake and xylem loading. Different from the BnBOR1;3a, the BnBOR1;1a and the BnBOR1;2 s showed the highest expression level in stems. This suggested that these genes were much more involved in B long distance transport, like the OsBOR1 in rice. Moreover, it was the first report that the BOR1-like genes were expressed in flower, which implied that these genes were involved in B translocation in reproductive development. The expression result suggested that the BnBOR1 s had undergone diversity in expression profile after that the B. napus diverged from A. thaliana.
Evidence of purifying selection under a divergent selective pressure
From the evolutionary viewpoint, the positive selection was used to calculate the ratio of the nonsynonymous substitution to the synonymous substitution (ω), where the ω > 1, = 1 and < 1 indicated positive selection, neutral evolution and negative selection, respectively [50, 51]. The branch models are useful for detecting the positive selection after gene duplications, where one copy of the duplicates with a new acquired function may have evolved at accelerated rates. In our analysis, the two-ratio model was run with the group III as the foreground branches to test whether these genes have on average evolved at a significantly different rate than the background branches, the other two groups, after duplication.
In our test with maximum likelihood estimates, the one-ratio model (M0) which averages ω (0.06159) over all sites and over all braches gave the results of ω < 1, revealing overall purifying selection acting over BnBOR1 s. LRTs indicated significant improvements in log likelihood values under the two ratios model (TR) relative to the M0 model for group III and the others (Table 4). The significantly better fit of the TR is consistent with a pattern of episodic positive selection acting in this lineage. They also support significant deviation of ω from 1. Estimated ω values under the fixed model (constrained to a single value across the lineage) were considerably less than 1 (0.09459 for the group III and 0.04768 for the others, respectively), which was consistent with predominantly purifying selection in the lineage. However, because purifying selection is ubiquitous and dominating in molecular evolution, positive selection most likely affects only a few sites [51, 52]. The site model analyses did not detect significant improvement in models (M8 vs M7) (Table 4), indicating that no positively selective sites were specific to group III (foreground lineage). However, six amino acids, 291 V, 386 K, 658L, 688A, 690C, 698S and 700L (numbering according to BnBOR1;1a), were identified by Bayes Empirical Bayes (BEB) analyses (Table 4, Supplementary material, Fig. S1) as candidates for positively selected sites, one with posterior probabilities >0.95 (700L). Instead, if we relaxed restriction on ω ≥ 1 but just allowed sites with different ω between the foreground and background lineages, then these sites were expected to evolve under divergent selective pressure. In this scenario, LRTs (M1a versus Clade model C) with group III as foreground provided significant rejection of null model (M1a), after Bonferroni’s correction (P < 0.001, df = 3). It suggested that the group III evolved under divergent selective pressure comparing with the others. Combined the tissue-specific expression, which Group III were ubiquitous expression but the others were not, the hypothesis was proposed that the difference of the tissue-specific expression put Group III under a divergent selective pressure. As discussed above, ω methods properly reflected the features of evolution of the BnBOR1 s genes in B. napus. This monitoring will help to understand the exiguous changes among lineages and further help to get insights into the nature of selective forces at molecular level in BnBOR1 s evolutionary processes.
Abbreviations
- B:
-
Boron
- MIPs:
-
Major intrinsic proteins
- RACE:
-
Rapid amplification of cDNA ends
- MYA:
-
Million years ago
- CDS:
-
Coding sequence
- PCR:
-
Polymerase chain reactions
- RT–PCR:
-
Reverse transcription polymerase chain reactions
- LRT:
-
Likelihood ratio test
- TR:
-
Two ration model
- HWSB:
-
Hot water-soluble boron
References
Warington K (1923) The effect of boric acid and borax on the broad bean and certain other plants. Ann Bot 37(4):629–672
Nielsen FH (2000) The emergence of boron as nutritionally important throughout the life cycle. Nutrition 16(7–8):512–514. doi:10.1016/S0899-9007(00)00324-5
Kobayashi M, Matoh T, Azuma J-i (1996) Two chains of rhamnogalacturonan II are cross-linked by borate-diol ester bonds in higher plant cell walls. Plant Physiol 110(3):1017–1020
Ishii T, Matsunaga T (1996) Isolation and characterization of a boron-rhamnogalacturonan-II complex from cell walls of sugar beet pulp. Carbohydr Res 284(1):1–9. doi:10.1016/008-6215(96)00010-9
O’Neill MA, Eberhard S, Albersheim P, Darvill AG (2001) Requirement of borate cross-linking of cell wall rhamnogalacturonan II for Arabidopsis growth. Science 294(5543):846–849. doi:10.1126/science.1062319
O’Neill MA, Ishii T, Albersheim P, Darvill AG (2004) Rhamnogalacturonan II: structure and function of a borate cross-linked cell wall pectic polysaccharide. Annu Rev Plant Biol 55:109–139. doi:10.1146/annurev.arplant.55.031903.141750
Marschner H (1995) Functions of mineral nutrients: micronutrients. In: Mineral nutrition of higher plants, 2nd ed. Academic Press, London, pp 313–404
Hu H, Brown PH, Labavitch JM (1996) Species variability in boron requirement is correlated with cell wall pectin. J Exp Bot 47(2):227–232. doi:10.1093/jxb/47.2.227
Raven JA (1980) Short- and long-distance transport of boric acid in plants. New Phytol 84:231–249
Brown PH, Shelp BJ (1997) Boron mobility in plants. Plant Soil 193(1):85–101
Dordas C, Brown PH (2000) Permeability of boric acid across lipid bilayers and factors affecting it. J Membrane Biol 175(2):95–105. doi:10.1007/s002320001058
Dannel F, Pfeffer H, Römheld V (2000) Characterization of root boron pools, boron uptake and boron translocation in sunflower using the stable isotopes 10B and 11B. Aust J Plant Physiol 27(5):397–405. doi:10.1071/PP99086
Stangoulis JC, Reid RJ, Brown PH, Graham RD (2001) Kinetic analysis of boron transport in Chara. Planta 213(1):142–146. doi:10.1007/s004250000484
Dordas C, Brown PH (2001) Evidence for channel mediated transport of boric acid in squash (Cucurbita pepo). Plant Soil 235(1):95–103. doi:10.1023/A:1011837903688
Dordas C, Chrispeels MJ, Brown PH (2000) Permeability and channel-mediated transport of boric acid across membrane vesicles isolated from squash roots. Plant Physiol 124(3):1349–1362
Nozawa A, Takano J, Kobayashi M, Wirén NV, Fujiwara T (2006) Roles of BOR1, DUR3, and FPS1 in boron transport and tolerance in Saccharomyces cerevisiae. FEMS Microbiol Lett 262(2):216–222. doi:10.1111/j.1574-6968.2006.00395.x
Takano J, Wada M, Ludewig U, Schaaf G, Wirén NV, Fujiwara T (2006) The Arabidopsis major intrinsic protein NIP5;1 is essential for efficient boron uptake and plant development under boron limitation. Plant Cell 18(6):1498–1509. doi:10.1105/tpc.106.041640
Tanaka M, Wallace IS, Takano J, Roberts DM, Fujiwara T (2008) NIP6;1 is a boric acid channel for preferential transport of boron to growing shoot tissues in Arabidopsis. Plant Cell 20(10):2860–2875. doi:10.1105/tpc.108.058628
Takano J, Noguchi K, Yasumori M, Kobayashi M, Gajdos Z, Miwa K, Hayashi H, Yoneyama T, Fujiwara T (2002) Arabidopsis boron transporter for xylem loading. Nature 420(6913):337–340. doi:10.1038/nature01139
Tanaka M, Fujiwara T (2008) Physiological roles and transport mechanisms of boron: perspectives from plants. Pflügers Archiv 456(4):671–677. doi:10.1007/s00424-007-0370-8
Takano J, Miwa K, Yuan L, Wirén NV, Fujiwara T (2005) Endocytosis and degradation of BOR1, a boron transporter of Arabidopsis thaliana, regulated by boron availability. Proc Natl Acad Sci USA 102(34):12276–12281. doi:10.1073/pnas.0502060102
Nakagawa Y, Hanaoka H, Kobayashi M, Miyoshi K, Miwa K, Fujiwara T (2007) Cell-type specificity of the expression of Os BOR1, a rice efflux boron transporter gene, is regulated in response to boron availability for efficient boron uptake and xylem loading. Plant Cell 19(8):2624–2635. doi:10.1105/tpc.106.049015
Sutton T, Baumann U, Hayes J, Collins NC, Shi B-J, Schnurbusch T, Hay A, Mayo G, Pallotta M, Tester M, Langridge P (2007) Boron-toxicity tolerance in barley arising from efflux transporter amplification. Science 318(5855):1446–1449. doi:10.1126/science.1146853
Miwa K, Takano J, Omori H, Seki M, Shinozaki K, Fujiwara T (2007) Plants tolerant of high boron levels. Science 318(5855):1417. doi:10.1126/science.1146634
Takano J, Toyoda A, Kasai K, Miwa K, Fuji K, Fujiwara T (2008) Endocytic degradation and polarized localization of borate transporters dependent on sorting motifs. In: 19th International Conference on Arabidopsis Research. Montreal, pp 197–198
Takano J, Tanaka M, Toyoda A, Miwa K, Kasai K, Fuji K, Onouchi H, Naito S, Fujiwara T (2010) Polar localization and degradation of Arabidopsis boron transporters through distinct trafficking pathways. Proc Natl Acad Sci USA 107(11):5220–5225. doi:10.1073/pnas.0910744107
Wolf BF, Wirén NV (2002) Plant biology: ping-pong with boron. Nature 420(6913):282–283. doi:10.1038/420282a
Park M, Li Q, Shcheynikov N, Zeng W, Muallem S (2004) NaBC1 is a ubiquitous electrogenic Na+-coupled borate transporter essential for cellular boron homeostasis and cell growth and proliferation. Mol Cell 16(3):331–341. doi:10.1016/j.molcel.2004.09.030
Kaya A, Karakaya HC, Fomenko DE, Gladyshev VN, Koc A (2009) Identification of a novel system for boron transport: Atr1 is a main boron exporter in yeast. Mol Cell Biol 29(13):3665–3674. doi:10.1128/MCB.01646-08
Franzke A, German D, Al-Shehbaz IA, Mummenhoff K (2009) Arabidopsis family ties: molecular phylogeny and age estimates in Brassicaceae. Taxon 58(2):425–437
U N (1935) Genome analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization. Jpn J Bot 7:385–452
Parkin IA, Sharpe AG, Lydiate DJ (2003) Patterns of genome duplication within the Brassica napus genome. Genome 46(2):291–303. doi:10.1139/g03-006g03-006
Wuding L (1995) Microelement nutrition and fertilization in china. China Agriculture Press, Beijing, pp 8–36
Yuai Y, Jianming X, Zhengqiang Y, Ke W (1993) Responses of rape genotypes to boron application. Plant Soil 155–156(1):321–324. doi:10.1007/BF00025047
Saghai-Maroof MA, Soliman KM, Jorgensen RA, Allard RW (1984) Ribosomal DNA spacer-length polymorphisms in barley: Mendelian inheritance, chromosomal location, and population dynamics. Proc Natl Acad Sci USA 81(24):8014–8018
Bork P, Dandekar T, Diaz-Lazcoz Y, Eisenhaber F, Huynen M, Yuan Y (1998) Predicting function: from genes to genomes and back. J Mol Biol 283(4):707–725. doi:10.1006/jmbi.1998.2144
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acid Symp Ser 41:95–98
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22(22):4673–4680
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24(8):1596–1599. doi:10.1093/molbev/msm092
Schmidt HA, Strimmer K, Vingron M, von Haeseler A (2002) TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 18(3):502–504
Yang Z (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24(8):1586–1591. doi:10.1093/molbev/msm088
Goldman N, Yang Z (1994) A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol 11(5):725–736
Yang Z (1998) Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol 15(5):568–573
Yang Z, Nielsen R, Goldman N, Pedersen AM (2000) Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155(1):431–449
Nielsen R, Yang Z (1998) Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148(3):929–936
Kyte J, Doolittle RF (1982) A simple method for displaying the hydropathic character of a protein. J Mol Biol 157(1):105–132. doi:10.1016/0022-2836(82)90515-0
Geourjon C, Deleage G (1995) SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput Appl Biosci 11(6):681–684
Connolly EL, Fett JP, Guerinot ML (2002) Expression of the IRT1 metal transporter is controlled by metals at the levels of transcript and protein accumulation. Plant Cell 14(6):1347–1357
Schaaf G, Schikora A, Haberle J, Vert G, Ludewig U, Briat JF, Curie C, von Wiren N (2005) A putative function for the arabidopsis Fe-Phytosiderophore transporter homolog AtYSL2 in Fe and Zn homeostasis. Plant Cell Physiol 46(5):762–774. doi:10.1093/pcp/pci081
Li WH, Gojobori T (1983) Rapid evolution of goat and sheep globin genes following gene duplication. Mol Biol Evol 1(1):94–108
Nei M (2005) Selectionism and neutralism in molecular evolution. Mol Biol Evol 22(12):2318–2342. doi:10.1093/molbev/msi242
Yang Z (2002) Inference of selection from multiple species alignments. Curr Opin Genet Dev 12(6):688–694. doi:10.1016/S0959-437X(02)00348-9
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (30771283, 30971861) and the National 863 High Technology Program of China (2007AA10Z117).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
11033_2011_930_MOESM1_ESM.png
{kind=link}
Supplementary figure Fig. S1 Multiple alignment of BnBOR1 s with AtBOR1 (NP_850469) Dots indicate identical residues with the BnBOR1;1a; grey box and star indicate possible positive selective sites detected by M8 model in PAml analysis; line indicates the conserved HCO3_cotransporer domain (PNG 160 kb)
Rights and permissions
About this article
Cite this article
Sun, J., Shi, L., Zhang, C. et al. Cloning and characterization of boron transporters in Brassica napus . Mol Biol Rep 39, 1963–1973 (2012). https://doi.org/10.1007/s11033-011-0930-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-011-0930-z