
Abstract
Pyruvate, an endogenous metabolite of glycolysis, is an anti-toxicity agent. Recent studies have suggested possible roles for pyruvate in protecting CNS neurons from excitotoxic and metabolic insults. Utilizing cultures derived from embryonic rat cortex, the studies presented in this paper indicate that an astroglia-mediated mechanism is involved in the neuroprotective effects of pyruvate against glutamate toxicity. Glutamate-induced toxicity could be reversed by pyruvate in a mixed culture of cortex cells. Importantly, in pure neuronal cultures from the same tissue, pyruvate failed to protect against glutamate toxicity. Addition of astroglia to the pure neuronal cultures restores the ability of pyruvate to protect neurons from glutamate-induced toxicity. Our results further suggest that pyruvate can induce glia to up-regulate the synthesis of glutathione (GSH), an antioxidant that protects cells from toxins such as free radicals. Taken together, our data suggest that astroglia in mixed cultures are essential for mediating the effects of pyruvate, revealing a novel mechanism by which pyruvate, an important intermediate of tricarboxylic acid cycle in the body, may act to protect neurons from damage during insults such as brain ischemia.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neurotoxicity in brain ischemia, traumatic brain injury and degenerative disease may be brought about by a chain of events involving failed energy metabolism, membrane depolarization, glutamate release, and toxic calcium influx. Of these, the excessive release of the excitatory neurotransmitter glutamate is considered to be the most damaging [1–3]. Glutamate-induced neurotoxicity involves mainly the activation of NMDA (N-methyl-d-aspartate) and Ca2+-permeable AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor) receptors [4, 5], which allow the influx of calcium and, consequently, the activation of a variety of molecules, including nitric oxide synthase (NOS) [6], xanthine oxidase [7], and mitogen-activated protein kinase [8]. Although antagonists to these glutamate receptors have shown promising protective effects against neuronal damage in experimental animal models of brain injury, these compounds failed to yield successful clinical results [9, 10]. Therefore, it is important to identify other neuroprotective agents that can avert glutamate-mediated neurotoxicity after acute brain injury.
Pyruvate, an endogenous metabolite of glycolysis, can work as an anti-toxicity agent and has been recently shown to have a protective role in CNS neurons, shielding them against excitotoxic and metabolic insults [11, 12]. However, the cellular location and the physiological significance of pyruvate have been the object of intense debate during the last decade. Previous studies have shown that pyruvate recycling takes place in the rat brain, with the cellular location being initially thought to be neuronal [13]. However, cell culture studies have pointed at astrocytes as the site of pyruvate recycling and release [14, 15]. Our current study aims to further investigate the protective roles of pyruvate in the CNS utilizing embryonic rat cortical cultures to demonstrate that pyruvate can act to protect neurons from glutamate-induced toxicity, and that astroglial cells play a central role in mediating the actions of pyruvate.
Our first set of experiments demonstrates that pyruvate blocks glutamate-induced neurotoxicity in mixed cultures. Most importantly, even when pyruvate is added after glutamate treatment, it can still act to protect neurons from cell death. However, addition of pyruvate to pure cortical neuronal cultures does not reduce neuronal loss after glutamate treatment, suggesting that other cell types present in the mixed culture are crucial for the protection by pyruvate. Additional studies employing pure astroglial cultures and co-culture of astroglia and neurons indicate that astroglia are essential for pyruvate’s action. The importance of astroglial cells was further confirmed by re-introducing these cells into pure neural cultures; this re-introduction restored the protective effects of pyruvate on neurons. Moreover, our data suggest that neuroprotection by pyruvate is through a combined action involving glutathione, which is the major cellular antioxidant and thiol compound, and which is known to play a central role in cellular antioxidative defense [16, 17].
In summary, our experiments demonstrate a novel action of pyruvate on the astroglial population in cortical cell cultures to protect neurons from glutamate toxicity.
Methods and materials
Materials
Long-Evans pregnant rats were obtained from the School of Medicine, Shanghai Jiaotong University (Shanghai, China). Culture medium and Hank’s solute were from GIBCO, Invitrogen Corporation. Reagents such as glutamate, pyruvate, l-Buthionine Sulfoximine (BSO), trypsin, fetal calf serum (FCS), standard GSH and GSSG were from Sigma Chemical Co., St. Louis, MO. Reagents used to treat cells were made as stock solutions. Glutamate and pyruvate were dissolved in Locke’s buffer (NaCl, 154 mM; KCl, 5.6 mM; CaCl2, 2.3 mM; MgCl2, 1.0 mM; NaHCO3, 3.6 mM; glucose, 5 mM; Hepes, 5 mM; pH 7.2) and diluted as indicated. Phenolsulfonphthalein-free MEM and the Alamar blue (AB) were purchased from Nalgene, BioSource, Intl, Int. U.S.A. All other chemicals were of the purest grade available from regular commercial sources.
Cultures and co-culture of cortical neurons and astrocytes
Cerebral cortices from 16 day old rat embryos (E16) were dissected and the meninges were removed. The tissue was gently triturated and enzymatically digested with 0.25% trypsin and 0.1 mg/ml DNase. The dissociated cells were suspended in the serum-containing medium (SCM: 89.4% MEM (Minimum Essential Medium; Gibco), 10% fetal calf serum, 0.6% glucose, supplemented with penicillin and streptomycin) for 6 days before treatment. To grow pure cortical neuronal cultures, SCM was changed to Neurobasal medium (NB) supplemented with B-27, penicillin, and streptomycin at 24 h after plating. After an additional 24 h, cytosine arabinoside (Ara-C, 5 μM) was added to these cultures for 3 days after which the Ara-C containing media was changed to fresh NB media. Cells were treated 24 h later. The culture media were always maintained at 37°C in a 5% CO2/95% air-humidified incubator.
Astrocyte cultures were prepared by a method modified from that described by McCarthy and de Vellis [18]. Cerebral cortices of 1 day old newborn rats (P1) were used. The tissue was enzymatically digested as described above. The dissociated cells were plated in poly-l-lysine precoated 75 cm2 plastic flasks at a high density of 0.6 × 106/cm2. The medium described above was changed twice a week. The cultures were grown to confluence in 2 weeks. At 14 days in vitro (DIV 14), the flasks were tightly sealed and shaken at 260 rpm for 18 h. The suspended cells in the flasks were discarded after shaking, whereas the adherent cells were flat, polygonal astrocytes, which were identified by morphology and by glial fibrillary acidic protein immunostaining. The density of confluent astrocytes was 0.1 × 106/cm2. Astrocyte-enriched cultures that were 2- to 6-week-old in flasks were used to make conditioned medium (CM).
To prepare CM, the SCM was added to the confluent astrocyte cultures at a concentration of 1.33 ml per 1 × 106/cm2 cells (10 ml per flask). After 3 days, pyruvate was added to some cultures, while other cultures were treated with Locke’s buffer (vehicle). The CM was collected from all cultures 24 h later. The different CMs were used in subsequent experiments to evaluate the possible involvement of pyruvate-elicited soluble factors in the observed effects.
To determine the contribution of direct astroglial cell presence to the neuroprotective effects of pyruvate, astroglial cultures were grown for 5 days in SCM before the cells were trypsinized and plated onto pure spinal cord neuronal cultures grown for 5 days in vitro (DIV 5). The co-culture was grown in SCM and treated 24 h later.
Alamar blue assay of neuronal viability and neuronal cell count
To assess cell viability, the Alamar blue (AB) assay was used as previously described [19, 20]. Culture medium was replaced with Phenolsulfonphthalein-free MEM containing 10% v/v Alamar blue and cultures were incubated for various periods at 37°C in humidified 95% air/5% CO2. Neuronal viability was measured when the medium in control wells turned from blue to pink for about 3 h. Then Alamar blue fluorescence was recorded in a spectrophotometric plate reader (DYNEX Technologies, USA), with excitation at 530 nm and emission at 590 nm.
At the same time, pictures were taken for 10 random fields in each experimental well, at a magnification of 100×. Cell number (as indicated by the appropriate immunostain) in each of the 10 pictures was estimated by an experimenter blind to the condition.
Fluorospectrophotometric assay of GSH/GSSG
The standard samples of reduction form glutathione (GSH) and oxidization form glutathione (GSSG) were scanned with full-wavelength range to determine the excitation wavelength: λex334.4 nm, the emission wavelength: λem424 nm, and the spectral bandwidth: 5 nm. Phosphate buffer saline (PBS) of pH 8.3 served as buffer solution. GSH was incubated for 30 min with 100 μl σ-phthaldehyde (OPA) of 10 mmol × l−1 methyl alcohol solution for derivatization, and then fluorescence intensities were measured. The GSH/GSSG ratio is commonly used to reflect the glutathione redox status.
Data analysis
The data are expressed as mean ± SD, and the differences between two groups were evaluated using Student’s t test. Multiple group comparisons were done using one-way analysis of variance (ANOVA) followed by the Dunn’s test. A probability level of 0.05 was used to establish significance.
Results
Pyruvate protects neurons from glutamate-induced neurotoxicity
Glutamate is the major physiological agent that mediates neuronal cell death. To examine the survival time of cortical neurons, we treated mixed cell cultures derived from embryonic day 16 (E16) rat brain with a range of concentrations of glutamate. These cells were grown in serum-containing medium (SCM) for 6 days (DIV 6) before being treated with various concentrations of glutamate for 1 h. After glutamate was removed, cells were kept in SCM for another 24 h before the cultures were fixed and immunostained. The number of surviving neurons was monitored with immunohistochemical labeling against MAP-2, a marker for neurons. The Alamar blue (AB) assay was used to assess cell viability.
As shown in Fig. 1, glutamate-elicited neuronal cell death increased in a dose-dependent manner. At high concentrations of glutamate (500 and 1,000 μM), approximately 50% of the neurons in these cultures underwent cell death.

Glutamate induces loss of cortical neurons in a dose-dependent manner. Cortical neurons were grown in SCM for 6 days before being treated with glutamate for l h. The numbers of neurons that survived were counted. Increasing concentrations of glutamate resulted in more significant loss of cortical neurons. Results were derived from three independent experiments (n = 7). * P < 0.05, ** P < 0.01 by nonparametric ANOVA followed by Dunn’s analysis for multiple comparisons contrasting glutamate-treated groups with control
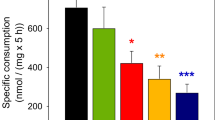
Additional experiments were designed to examine whether pyruvate could reduce glutamate-induced neuronal cell death. Cells were treated with 500 μM glutamate in the presence of various concentrations of pyruvate (0–1 mM). Each concentration of pyruvate was applied either together with glutamate (Fig 2a), or following the removal of glutamate (Fig 2b). As shown in Fig. 2, pyruvate blocked glutamate toxicity in a dose-dependent manner. At high concentrations of pyruvate, glutamate-promoted neuronal cell death was completely abolished (Fig. 2a). Most importantly, treatment with pyruvate after the termination of glutamate exposure resulted in similar neuroprotection as with concomitant application (Fig. 2b). These results suggest that pyruvate can act to protect neurons after glutamate exposure in culture.

Pyruvate blocks glutamate toxicity to neurons. Cortical neurons were grown in SCM for 6 days. Then they were treated with glutamate for l h with various concentrations of pyruvate (0–l mM). a When glutamate and pyruvate were added together, pyruvate blocked glutamate toxicity. Results were derived from five independent experiments (n = 6). b When pyruvate was added after glutamate treatment, it yielded similar protective effects as in A. Results were derived from five independent experiments (n = 7). * P < 0.05, ** P < 0.01 by nonparametric ANOVA followed by Dunn’s analysis for multiple comparisons using vehicle as control
Pyruvate cannot protect neurons from glutamate toxicity in pure neuronal cultures
To address the question of whether the neuroprotective effects of pyruvate are mediated directly by neurons or indirectly by other cortical cells, pure cortical neuronal cultures were established. Twenty-four hours after plating in SCM, SCM was changed to NB medium, which optimizes neuronal growth. Ara-C (51 M) was added to the cultures after another 24 h period to eliminate glial populations. The medium was changed after 3 days, and cells were treated 24 h later. This procedure eliminated non-neuronal cell types in the cultures, while allowing the survival of neurons.
Dose response experiments demonstrated that these cultures were much more sensitive to glutamate toxicity. Treatment with 10 μM glutamate resulted in very significant neuronal loss (>80%; Fig. 3a), and 500 μM glutamate eliminated almost all neurons. The effect of this second concentration is particularly remarkable when compared to the neuronal loss observed in mixed cultures treated with the same concentration of glutamate (only 50%; Fig. 1).

Neuroprotection by pyruvate against glutamate neurotoxicity is mediated by astrocytes. a Dose-dependent neurons loss in pure cortical neuron cultures due to glutamate toxicity. Pure cortical neurons were established, and after 6 days, these cells were treated with glutamate for l h. The number of neurons that survived were counted. Increasing concentration of glutamate resulted in a more significant loss of neurons. b Pyruvate does not decrease glutamate toxicity in pure cortical neuron cultures. Six days after plating, pure cortical neuron cultures were treated with glutamate (10 or 500 μM) for l h. Pyruvate (0.25 or 0.75 mM) was added after glutamate treatments. Cells were fixed after 24 h and numbers of survival neurons were counted. c Re-plating of astroglial cells in pure neuron cultures reinstates the effects of pyruvate. Pure astroglial cells cultures were established, then trypsinized and re-plated in pure neuron cultures grown in SCM for 5 days. After 24 h, cells were exposed to glutamate (10 or 500 μM) for l h and pyruvate (0.25 or 0.75 mM) was added after the medium was changed. The number of neurons that survived were counted after 24 h. Results were derived from three independent experiments (n = 7). * P < 0.05, ** P < 0.01 by nonparametric ANOVA followed by Dunn’s analysis comparing glutamate treated with control
To examine whether pyruvate can directly reduce glutamate toxicity in these cultures, cells were treated with various concentrations of pyruvate either concurrently with, or at the termination of, the application of 10 or 500 μM glutamate. Pyruvate itself had no effect on neuronal survival. In addition, in these conditions, pyruvate conveyed no significant protection against glutamate toxicity (Fig. 3b). Even high concentrations of pyruvate (up to 1 mM) failed to affect the neuronal loss elicited by the smallest dose of glutamate tested (10 μM). These results suggest that the protective effects of pyruvate are mediated by cortical cells other than neurons.
Astroglia play an important role in mediating the effects of pyruvate
One candidate cell type that could mediate the effects of pyruvate are the astroglial populations, which have been reported to protect neurons from excitotoxic insults in cases of CNS trauma or brain ischemia. Therefore, our next step was to examine whether astroglia contribute to the effects of pyruvate.
To establish pure cortical astroglial cultures, cells from P1 rat brains were plated and grown in high serum conditions for 9 days before undergoing sequential shaking procedures to remove microglia and oligodendrocytes. After three more days in Ara-C supplemented medium, cells were replated. At this stage, there were neither neurons nor oligodendrocytes present in these cultures.
Our studies then explored whether direct addition of astroglia to pure neuronal cultures could restore the protective effects of pyruvate against glutamate toxicity. These cells were grown for 5 days in SCM and then trypsinized and replated onto DIV 5 pure cortical neuronal cultures. The medium for the combined cultures was changed to SCM. Twenty-four hours later, the combined cultures were treated with glutamate followed by the addition of pyruvate or vehicle. Examination of numbers of surviving neurons indicated that both 0.25 and 0.75 mM pyruvate blocked the toxic effects of 10 and 500 μM glutamate (Fig. 3c), suggesting that astroglia is essential for mediating the protective effects of pyruvate.
Astroglia-mediated neuroprotection by pyruvate may involve GSH
To further elucidate the neuroprotective mechanisms of pyruvate, we turned our attention to Glutathione (GSH), an antioxidant that is known to protect neurons from toxins such as free radicals. In this next set of experiments, Buthionine Sulfoximine (BSO), an inhibitor of the restriction enzyme Glucosylceramide Synthase (GCS) that is necessary for the synthesis of GSH, was been added to the culture medium.
Combined cultures (neurons and astroglia) were treated with glutamate followed by the addition of either 0.5 mM pyruvate or 0.1 mM BSO. Estimations of the number of surviving neurons indicated that while BSO had no direct effect on neuronal survival, it was able to block the protective effects of pyruvate (Fig. 4a). These results strongly suggest that GSH plays an important role in pyruvate-mediated protection from glutamate excitotoxicity.

Neuroprotective effect of pyruvate relates to the synthesis of GSH. a BSO inhibit the neuroprotective effect of pyruvate. Cortical neurons were grown in SCM for 6 days before treated with glutamate (500 μM) in the presence or absence of pyruvate (0.5 mM) of BSO (0.1 mM). When glutamate, pyruvate and BSO were added together. BSO inhibit the effect of pyruvate. b Pyruvate increases the synthesis of GSH and elevates the ratio of GSH:GSSG The combined cultures were treated with glutamate (500 μM) followed by the addition of pyruvate (0.5 mM), BSO (0.1 mM) or pyruvate + BSO. Fluorospectrophotometric assay was used to detect the GSH and GSSG in the CM. The results of GSH/GSSG were calculated. Results were derived from three independent experiments (n = 5). ** P < 0.01 by nonparametric ANOVA followed by Dunn’s analysis comparing glutamate treated with control
A fluorospectrophotometric assay was used to detect GSH and GSSG in the CM. The GSH/GSSG ratio has been used to reflect the glutathione redox status. BSO significantly inhibited GSH synthesis (Fig. 4b). However, 0.5 mM pyruvate could promote GSH synthesis and inhibit the production of GSSG. So, pyruvate can elevate the GSH/GSSH ratio significantly, an effect that could be involved in pyruvate-mediated protection from glutamate toxicity.
Discussion
The results of our study indicate that an astroglia-dependent mechanism is involved in mediating the protective effects of pyruvate on neurons against glutamate toxicity. While glutamate-induced neurotoxicity can be reversed by treatment with pyruvate in a mixed culture of cortex cells, pyruvate has no protective effect when applied to pure neuronal cultures from the same tissue. Addition of astroglia to the pure neuronal cultures restores the ability of pyruvate to protect the neurons from glutamate-induced toxicity. Our results also suggest that astroglia can up-regulate the synthesis of GSH, which is an antioxidant that protects neurons from toxins such as free radicals.
In our experiments, cells were exposed to a brief 1 h treatment with glutamate. The putative neuroprotective role of pyruvate was tested using 500 μM glutamate. In brain cultures grown in SCM, we demonstrated that pyruvate protected cortical neurons against glutamate toxicity. A concentration of 0.5 μM pyruvate reversed the cell loss elicited by treatment with 500 μM glutamate (Fig. 2). It should be noted that cultures grown in SCM are a mixed population; not only neurons, but also oligodendrocytes and astroglial cells are present in these cultures. To evaluate whether oligodendrocytes and astroglia were involved in the protective actions of pyruvate, pure cortical neuronal cultures were developed. Interestingly, pyruvate did not significantly protect cortical neurons against glutamate toxicity in these cultures (Fig. 3b). The lack of effects by pyruvate on pure neurons suggests that glial cells are essential for reducing the damage to neurons in the CNS.
Pyruvate is an anti-toxicity agent that is an endogenous metabolite of glycolysis. Many data have suggested that pyruvate can protect neurons against a variety of insults, such as hydrogen peroxide-induced toxicity [21, 22], cystein’s neurotoxicity [23], and zinc-induced neuronal death [24]. Recently, Lee [12] reported the protection by pyruvate against transient forebrain ischemia in rats. However, this study failed to find a similar protective effect against the glutamate-NMDA-mediated neurotoxicity in an in vitro test. In contrast, our study showed that 1 mM pyruvate in culture medium was able to reduce the degree of glutamate neurotoxicity. The reason for the discrepancy between these two studies may be that the different models of the glutamate-induced neurotoxicity used. While in Lee’s test glutamate was present in the culture medium for 24 h, in our experiment, pyruvate was added after 30 min of glutamate treatment, which is likely to have improved neuronal survival. Maus [25] found that pyruvate and lactate could protect striatal neurons against NMDA-induced neurotoxicity. However, the toxic effects on the neurons in their study were mediated by the generation of oxygen free radicals and also related to activity. They did not test whether pyruvate could counteract glutamate-mediated neurotoxicity. Like other types of keto acids, pyruvate can react with hydrogen peroxide through a non-enzymatic reaction, and it can function as an oxygen free radical scavenger. As a result, we speculated that the mechanism of neuroprotection by pyruvate against glutamate-induced neurotoxicity could be related to the protection of neurons from hydrogen peroxide.
In order to further clarify this, we examined one kind of antioxidant system in astrocyte cells: glutathione (GSH) [26, 27]. Our results showed that GSH is indeed involved in the pyruvate-induced protection against glutamate neurotoxicity. The synthesis of GSH was inhibited by BSO, an inhibitor of GCS, a key enzyme in the synthesis of GSH. Specifically, BSO is an amino acid analog that inhibits the synthesis of glutathionylcysteine and thus of glutathione (Glu-Cys-Gly:GSH). BSO is phosphorylated in the active site of this enzyme, and irreversible inhibition results from the noncovalent but tight binding of buthionine sulfoximine phosphate. This inhibition occurs in a dose- and time-dependent manner. We found that low concentrations of BSO did not significantly increase neuronal death. However, the neuroprotective effect of pyruvate was inhibited when BSO and pyruvate were simultaneously added to the neuronal culture medium. It is unlikely that the addition of BSO could lead to a synergistic effect that could induce the death of neurons. Therefore, this result strongly suggests that the neuroprotection of pyruvate against glutamate-mediate neurotoxicity involves the participation of the GSH. The exact mechanism remains to be elucidated and future studies, both in vitro and in vivo, will likely provide further insights into the interaction of pyruvate with astrocytes and the resulting effect on neuronal survival. Nevertheless, we believe that neuroprotective efforts targeting the functional integrity of astrocytes may constitute a superior strategy for improving neuroprotection in the event of insults to the CNS.
Conclusion
Our study showed that astroglia in mixed cultures are essential for mediating the neuroprotective effects of pyruvate. These results reveal a novel mechanism by which pyruvate, an important intermediate of the tricarboxylic acid cycle in the body, may act to protect neurons from damage during insults such as brain ischemia. Furthermore, we also found that astroglia-mediated neuroprotection by pyruvate may involve GSH and be related to its antioxidant effects.
References
Magistretti PJ, Pellerin L, Rothman DL, Shulman RG (1999) Energy on demand. Science 283:496–497
Coyle JT, Puttfarcken P (1993) Oxidative stress, glutamate, and neurodegenerative disorders. Science 262:689–695
Lipton SA, Rosenberg PA (1994) Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med 330:613–622
Urushitani M, Nakamizo T, Inoue R et al (2001) N-methyl-d-aspartate receptor-mediated mitochondrial Ca(2+) overload in acute excitotoxic motor neuron death: a mechanism distinct from chronic neurotoxicity after Ca(2+) influx. J Neurosci Res 63:377–387
Matthews CC, Zielke HR, Wollack JB, Fishman PS (2000) Enzymatic degradation protects neurons from glutamate excitotoxicity. J Neurochem 75:1045–1052
Sanelli T, Strong MJ (2007) Loss of nitric oxide-mediated down-regulation of NMDA receptors in neurofilament aggregate-bearing motor neurons in vitro: implications for motor neuron disease. Free Radic Bio Med 42:143–151
Sanganahalli BG, Joshi PG, Joshi NB (2005) Xanthine oxidase, nitric oxide synthase and phospholipase A2 produce reactive oxygen species via mitochondria. Brain Res 1037:200–203
Eftmova T, Deucher A, Kuroki T, Ohba M, Eckert RL (2002) Novel protein kinase C isoforms regulate human keratinocyte differentiation by activating a p38 delta mitogen-activated protein kinase cascade that targets CCAAT/enhancer-binding protein alpha. J Bio Chem 277:31753–31760
Ginsberg MD (2009) Current status of neuroprotection for cerebral ischemia. Synoptic overview. Stroke 40:111–114
Trendelenburg G, Dirnagl U (2005) Neuroprotective role of astrocytes in cerebral ischemia: focus on ischemic preconditioning. Glia 50:307–320
Desagher S, Glowinski J, Premont J (1997) Pyruvate protects neurons against hydrogen peroxide-induced toxicity. J Neurosci 17:9060–9067
Lee JY, Kim YH, Koh JY (2001) Protection by pyruvate against transient forebrain ischemia in rats. J Neurosci 21:RC171
Olstad E, Olsen GM, Qu H, Sonnewald U (2007) Pyruvate recycling in cultured neurons from cerebellum. J Neurosci Res 85:3318–3325
Alves PM, Nunes R, Zhang CH et al (2000) Metabolism of 3-C-13-malate in primary cultures of mouse astrocytes. Dev Neurosci 22:456–462
Waagepetersen HS, Qu H, Hertz L, Sonnewald U, Schousboe A (2002) Demonstration of pyruvate recycling in primary cultures of neocortical astrocytes but not in neurons. Neurochem Res 27:1431–1437
Frenzel J, Richter J, Eschrich K (2005) Pyruvate protects glucose-deprived miffler cells from nitric oxide-induced oxidative stress by radical scavenging. Glia 52:276–288
Kala G, Hertz L (2005) Ammonia effects on pyruvate/lactate production in astrocytes—interaction with glutamate. Neurochem Int 47:4–12
McCarthy KD, Devellis J (1980) Preparation of separate astroglial and oligodendrogial cell-cultures from rat cerebral tissue. J Cell Biol 85:890–902
Goodwin GW, Kuntz MJ, Paxton R, Harris RA (1987) Enzymatic determination of the branched-chain alpha-keto acids. Ana Biochem 162:536–539
White MJ, DiCaprio MJ, Greenberg DA (1996) Assessment of neuronal viability with Alamar blue in cortical and granule cell cultures. J Neurosci Meth 70:195–200
Wang XF, Cynader MS (2001) Pyruvate released by astrocytes protects neurons from copper-catalyzed cysteine neurotoxicity. J Neurosci 21:3322–3331
Wang XF, Cynader MS (1999) Effects of astrocytes on neuronal attachment and survival shown in a serum-free co-culture system. Brain Res Protoc 4:209–216
Nakamichi N, Kambe Y, Oikawa H et al (2005) Protection by exogenous pyruvate through a mechanism related to monocarboxylate transporters against cell death induced by hydrogen peroxide in cultured rat cortical neurons. J Neurochem 93:84–93
Lobner D, Canzoniero LM, Manzerra P et al (2000) Zinc-induced neuronal death in cortical neurons. Cell Mol Biol 46:797–806
Maus M, Marin P, Israel M, Glowinski J, Premont J (1999) Pyruvate and lactate protect striatal neurons against N-methyl-d-aspartate-induced neurotoxicity. Eur J Neurosci 11:3215–3224
Regan RF, Guo Y (1999) Extracellular reduced glutathione increases neuronal vulnerability to combined chemical hypoxia and glucose deprivation. Brain Res 817:145–150
Ramírez BG, Rodrigues TB, Violante IR et al (2007) Kinetic properties of the redox switch/redox coupling mechanism as determined in primary cultures of cortical neurons and astrocytes from at brain. J Neurosci Res 85(15):3244–3253
Acknowledgments
This study was funded by grants from the Scientific Research Foundation of Nanjing Medical University (No. 2002-19) and the Shanghai Science and Technology Committee (No. 074107019).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Miao, Y., Qiu, Y., Lin, Y. et al. Protection by pyruvate against glutamate neurotoxicity is mediated by astrocytes through a glutathione-dependent mechanism. Mol Biol Rep 38, 3235–3242 (2011). https://doi.org/10.1007/s11033-010-9998-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-010-9998-0