
Abstract
The objective of the work was to study PIK3CA mutations in wild type KRAS and BRAF colorectal cancer. Clinicopathological data and paraffin-embedded specimens were collected on 73 patients who underwent colorectal resections at General Yagüe Hospital in Burgos. KRAS, BRAF and PIK3CA status were analyzed by real-time PCR in all patients. PIK3CA mutations were present in 8.22% of wild type KRAS and BRAF colorectal cancers. The most frequent mutation is E545K/D in exon 9 which represents 83.3% of all mutations. By contrast, we did not found any tumour harbouring H1047R mutation in exon 20. Among the patients who undergo a curative resection of colorectal cancer, PIK3CA mutation is present in an important percentage of KRAS and BRAF wild type tumours. PIK3CA mutation may be considered as it could be a hypothetic reason to be not responder to anti-EGFR antibodies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer is the second commonest cause of cancer-related death in the United Sates and Western World. In the last few years its incidence has increased while the age at diagnosis is decreasing [1]. New therapeutic options have increased the overall survival rate of advanced disease from 10 to 18–24 months during the past decade [2]. Advances in surgery, chemotherapy and adjuvant therapy are effective in prolonging time to disease progression and survival in patients with advanced colorectal cancer [3]. Changes in adjuvant therapy include treatment with oxaliplatin combined with 5-FU/leucovorin or capecitabine, with the use concomitant of targeted agents such as cetuximab and bevacizumab.
Epidermal growth factor receptors (EGFRs) have been validated as a therapeutic target in colorectal cancer (CRC) [4, 5]. Ligand occupancy of the EGFR activates the RAS/RAF/MAPK, STAT, and PIK3/AKT signalling pathways, which together modulate cellular proliferation, adhesion, angiogenesis, migration, and survival [6, 7].
KRAS and BRAF can harbour oncogenic mutations that yield a constitutively active protein [8–10]. Such mutations are found in approximately 30–50% [11, 12] and 10–15% [12, 13] of CRC tumours, respectively. Several studies have indicated that the presence of mutant KRAS in CRC tumours correlates with poor prognosis [12, 13] and is associated with lack of response to EGFR inhibitors [14–17].
In several studies wild type KRAS and BRAF status was shown to be required but not sufficient to confer sensitivity to anti-EGFR antibodies as cetuximab and panitumumab. The mechanisms of resistance to these drugs in patients with wild type KRAS and BRAF tumours are unknown. One potential mechanism making the patients without mutations in KRAS and BRAF fails to respond to anti-EGFR therapy could be mutations in PIK3.
Activation of the Phosphatidylinositol 3-kinase PIK3/AKT pathway is thought to play a critical role in the development of a variety of human malignancies [18–20]. The PIK3CA gene encodes the catalytic subunit p110α of PIK3. Mutant PIK3CA stimulates the AKT pathway and promotes cell growth in several cancers, including colorectal cancer being associated in these cases with poor prognosis [21].
Elucidating mechanisms of resistance to these antibodies will prove to be important for the selection of therapeutic combinations in order to maximize clinical benefit.
In addition to ascertaining resistance mechanisms, the study of biomarkers such PIK3CA mutations will be useful to further refine the responder population and would allow to offer to patients other alternative treatments.
In colorectal cancer there are only a small number of studies analyzing that and numerous discrepancies in the PIK3CA mutations frequencies reported ranking from 10 to 30% [22–24]. Thus the percentage and distribution of PIK3CA mutations in colorectal cancer still remains uncertain and this retrospective study which is the first in Spain, to our knowledge will help to clarify.
The aim of this study was to evaluate a cohort of 73 wild type KRAS and BRAF CRC patients in order to investigate the percentage of them which harbours PIK3CA mutations and it could be a hypothetic reason for being non-responder to anti-EGFR therapies.
Materials and methods
Patients
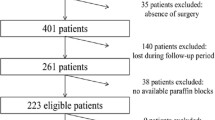
This retrospective study included 73 KRAS and BRAF wild type patients with histological confirmed colorectal primary tumour at the Pathology Department in General Yagüe Hospital (Burgos, Spain). Clinical data were obtained from the tumour registry and from hospital charts. These data included patient age at diagnosis, gender, Dukes Stage and Histological grade (see Table 1).
Assays methods
Mutational analysis of KRAS, BRAF and PIK3CA in tumor samples
Formalin-fixed, paraffin-embedded tumour sections were deparaffinized and air dried, and DNA was isolated using proteinase K and QIAamp® DNA FFPE Tissue kit (Qiagen) according to the manufacturer’s protocol.
KRAS
Mutant KRAS in exon 2 was detected using a validated KRAS mutation kit (DxS Ltd, Manchester, United Kingdom) and following manufacturer’s instructions. This analysis identifies seven somatic mutations located in codons 12 and 13 (Gly12Asp, Gly12Ala, Gly12Val, Gly12Ser, Gly12Arg, Gly12Cys, and Gly13Asp) using allele-specific real-time polymerase chain reaction [25–27]. The analysis was performed in an ABI Prism 7500 instrument (Applied Biosystems).
BRAF
BRAF mutation in exon 15 and codon 600 (V600E) were evaluated modifing a method previously reported by Benlloch et al. [28]. The primers were designed to avoid amplification of a pseudogen located in chromosome X. Briefly, we used the set of primers and probes as follow: BRaf-F (forward) 5′CTACTGTTTTCCTTTACTTACTACACCTCAGA-3′: BRaf-R (reverse) 5′ATCCAGACAACTGTTCAAACTGATG-3′, Wild type probe 5′VICCTAGCTACAGtGAAATC-3′ and Mutant probe 5′-FAM-TAGCTACAGaGAAATC-3′. The primers and probes were tested with controls of DNA from the HT29 cell line (ATCC) which harbours the V600E B-Raf heterozygotic mutation and DNA from the SW480 cell line (ATCC) which is BRAF wild type and KRAS mutant. Real-time PCR was performed in a final reaction volume of 20 μl containing 10 μl of 2× Genotyping PCR Master Mix (Applied Biosystems), 900 nmol/l of each primer, 250 nmol/l of each probe, and 5 μl of DNA solution. PCR was performed in MicroAmp optical 96-well plates with optical adhesive covers (Applied Biosystems). Amplification and detection were performed with an ABI prism 7500 sequence detection system (Applied Biosystems). The amplification conditions were 2 min at 50°C for AmpErase uracil-N-glycosylase activity and 10 min at 95°C for AmpliTaq Gold activation, followed by 50 cycles of 15 s at 92°C for denaturation and 1.5 min at 60°C for annealing and extension. The fluorescence data were analyzed with the allelic discrimination software of the ABI Prism 7500 instrument.
PIK3CA
PIK3CA mutations in exons 9 and 20 were detected using a validated PIK3CA mutation kit (DxS Ltd, Manchester, United Kingdom) that identifies four somatic mutations (H1047R, E542K, E545D and E545K) using Real-Time Polymerase Chain Reaction based in ARMS® and Scorpion® technology. This method is highly selective and can detect approximately 1–2% mutants in a background of genomic DNA [29]. The analysis was performed in an ABI Prism 7500 instrument (Applied Biosystems).
Statistical analysis methods
Statistical analysis was performed using SPSS version 17.0. A comparison has been made among the variables. We used Chi-square distribution on categorical variables and the non parametrical U-Mann–Whitney distribution on continuous variables and in both cases P < 0.05 was considered to be statistically significant.
Results and discussion
PIK3CA mutational profile
We analyzed 73 patients (71.3% male and 28.7% female) aged 31 to 88 years old, medium age 69.9. Six of 73 successfully analysed samples (8.22%) carried a PIK3CA mutation. The missense mutations were found in the hot spots located in exon 9 of the PIK3CA gene (E542D, E545D, E545K) (Table 2) while we did not found any mutation in exon 20 (H1047R).
We observed one tumour with E542K mutation and five with E545K/D mutation. In our population, the most frequent mutation is E545K/D which represents 83.3% of all mutations. By contrast, we did not found any tumour harbouring H1047R mutation in exon 20.
Making a comparison between the age and the PIK3CA mutations using Mann–Whitney test we found that there is significant difference between age and PIK3CA mutations (signification of 0.026 and sample power 47%). Comparing sex and PIK3CA mutations using Chi-square test we found no significant difference between male and female (signification of 0.999 and sample power 9%).
In a Spanish population we found a distribution of specific PIK3CA mutations similar to Perrone’s data [24] where mutations in exon 9 were the most frequent and no mutation in exon 20 was found. We speculate that the specific distribution of PIK3CA mutations could be homogeneous in different populations as the Spanish cohort is similar to the European series published although both published series have a relatively small sample size and future studies are required to confirm these data.
There are numerous discrepancies in the PIK3CA gene mutation frequencies reported from a small number of studies and previous data reported different percentages ranking from 10 to 30% (including KRAS and BRAF wild type and mutant) [22–24, 30].
This retrospective study, analyze PIK3CA mutation in wild type KRAS and BRAF patients, has shown that there is an important percentage of these tumours which have PIK3CA/AKT deregulation.
We found an 8.22% of patients who present PIK3CA mutations. Our results are similar to Velho et al. [31] who determined that the prevalence of PIK3CA mutations in 51 CRC patients was 7.1.
These results are important because corroborate the data presented by Ogino et al. and Baldus et al. [32] describing that in KRAS wild type tumours, the presence of PIK3CA mutations was associated with both a bad prognosis and with an increase in colorectal cancer-specific mortality.
We have demonstrated compelling evidence supporting the PIK3CA mutation in colorectal carcinoma. Although we have not provided data about patients’ response, our data attend to speculate that futures studies are necessary to prove that some patients who present PIK3CA could be non-responders to anti-EGFR therapies as cetuximab.
Therefore, as we have demonstrated that PIK3CA gene is mutated in an important percentage of KRAS and BRAF wild type patients, we thought that this could be a potential target for developing drugs to treat colorectal cancer.
Conclusion
PIK3CA mutations may be one of the genes that could predict the lack of efficacy of EGFR inhibitors in KRAS and BRAF patients.
PIK3CA mutation, observed in more than 8% of the KRAS and BRAF wild type patients, could predict escape from cetuximab treatment. These results also suggest that PIK3CA could be a promising target for adjuvant therapy in colorectal cancer patients.
Abbreviations
- BRAF :
-
V-raf murine sarcoma viral oncogene homolog B1
- KRAS :
-
Human homolog of the Kirsten rat sarcoma-2 virus oncogene
- EGFR:
-
Epidermal grown factor receptor
- CRC:
-
Colorectal cancer
- PIK3 :
-
Phosphatidylinositol 3-kinase
References
Jemal A, Siegel R, Ward E et al (2008) Cancer statistics, 2008. CA Cancer J Clin 58(2):71–96
Waldner MJ, Neurath MF (2010) The molecular therapy of colorectal cancer. Mol Aspects Med 31(2):171–178
Meulenbeld HJ, Creemers GJ (2007) First-line treatment strategies for elderly patients with metastatic colorectal cancer. Drugs Aging 24(3):223–238
Cunningham D, Humblet Y, Siena S et al (2004) Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med 351(4):337–345
Van Cutsem E, Peeters M, Siena S et al (2007) Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol 25(13):1658–1664
Hynes NE, Lane HA (2005) ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 5(5):341–354
Mendelsohn J, Baselga J (2006) Epidermal growth factor receptor targeting in cancer. Semin Oncol 33(4):369–385
Downward J (2003) Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 3(1):11–22
Malumbres M, Barbacid M (2003) RAS oncogenes: the first 30 years. Nat Rev Cancer 3(6):459–465
Schubbert S, Shannon K, Bollag G (2007) Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer 7(4):295–308
Amado RG, Wolf M, Peeters M et al (2008) Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 26(10):1626–1634
Laurent-Puig P, Cayre A, Manceau G et al (2009) Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J Clin Oncol 27(35):5924–5930
Di Nicolantonio F, Martini M, Molinari F et al (2008) Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol 26(35):5705–5712
Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F et al (2007) Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res 67(6):2643–2648
De Roock W, Piessevaux H, De Schutter J et al (2008) KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann Oncol 19(3):508–515
Di Fiore F, Blanchard F, Charbonnier F et al (2007) Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by cetuximab plus chemotherapy. Br J Cancer 96(8):1166–1169
Lievre A, Bachet JB, Le Corre D et al (2006) KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res 66(8):3992–3995
Engelman JA, Luo J, Cantley LC (2006) The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev 7(8):606–619
Karakas B, Bachman KE, Park BH (2006) Mutation of the PIK3CA oncogene in human cancers. Br J Cancer 94(4):455–459
Manning BD, Cantley LC (2007) AKT/PKB signaling: navigating downstream. Cell 129(7):1261–1274
Samuels Y, Velculescu VE (2004) Oncogenic mutations of PIK3CA in human cancers. Cell Cycle 3(10):1221–1224
Ikenoue T, Kanai F, Hikiba Y et al (2005) Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res 65(11):4562–4567
Ogino S, Nosho K, Kirkner GJ et al (2009) PIK3CA mutation is associated with poor prognosis among patients with curatively resected colon cancer. J Clin Oncol 27(9):1477–1484
Perrone F, Lampis A, Orsenigo M et al (2009) PI3KCA/PTEN deregulation contributes to impaired responses to cetuximab in metastatic colorectal cancer patients. Ann Oncol 20(1):84–90
Newton CR, Graham A, Heptinstall LE et al (1989) Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS). Nucleic Acids Res 17(7):2503–2516
Thelwell N, Millington S, Solinas A, Booth J, Brown T (2000) Mode of action and application of scorpion primers to mutation detection. Nucleic Acids Res 28(19):3752–3761
Whitcombe D, Theaker J, Guy SP, Brown T, Little S (1999) Detection of PCR products using self-probing amplicons and fluorescence. Nat Biotechnol 17(8):804–807
Benlloch S, Paya A, Alenda C et al (2006) Detection of BRAF V600E mutation in colorectal cancer: comparison of automatic sequencing and real-time chemistry methodology. J Mol Diagn 8(5):540–543
Board RE, Thelwell NJ, Ravetto PF et al (2008) Multiplexed assays for detection of mutations in PIK3CA. Clin Chem 54(4):757–760
Ekstrand AI, Jonsson M, Lindblom A, Borg A, Nilbert M (2010) Frequent alterations of the PI3K/AKT/mTOR pathways in hereditary nonpolyposis colorectal cancer. Fam Cancer 9(2):125–129
Velho S, Oliveira C, Ferreira A et al (2005) The prevalence of PIK3CA mutations in gastric and colon cancer. Eur J Cancer 41(11):1649–1654
Baldus SE, Schaefer KL, Engers R, Hartleb D, Stoecklein NH, Gabbert HE (2010) Prevalence and heterogeneity of KRAS, BRAF, and PIK3CA mutations in primary colorectal adenocarcinomas and their corresponding metastases. Clin Cancer Res 16(3):790–799
Acknowledgments
This work was supported by a grant FIS CA08/00070 from Instituto de Salud Carlos III, Spanish Ministerio de Ciencia e Innovación to MHV, Caja de Burgos Fundación Burgos por la Investigación de la Salud and Junta de Castilla y León. MHV is especially thankful to CVP, IHH and AHV, for their support.
Conflict of interest statement
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Herreros-Villanueva, M., Gomez-Manero, N., Muñiz, P. et al. PIK3CA mutations in KRAS and BRAF wild type colorectal cancer patients. A study of Spanish population. Mol Biol Rep 38, 1347–1351 (2011). https://doi.org/10.1007/s11033-010-0236-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-010-0236-6