
Abstract
Osteoarthritis (OA) is a most common multifactorial degenerative joint disease in elderly individuals. OA is affecting severely the quality of life of patients, while the causes of OA are not completely understood. Age, obesity, the female sex, and previous injury are considered as significant risk factors. Recently, increased levels of adipokines which are mainly produced by adipocytes have been detected in patients with osteoarthritis. Moreover, studies on different adipokines all reveal that they have played proinflammatory and catabolic/anabolic roles during the pathophysiology of OA. In the present review, we summarize current data on the effect of the adipose tissue-derived hormones leptin, adiponectin, resistin and visfatin on initiation and progression of OA.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Osteoarthritis (OA), a degenerative joint disorder, is characterized by degeneration of articular cartilage, changes in subchondral bone, osteophyte formation, and synovial inflammation. It is the most common arthritis and may lead to severe symptoms like pain, malformation of the joint and disability [1], while the causes of which are poorly understood. Present studies have proven the age, obesity, female sex and previous injury may play an important role in the process of OA [2].
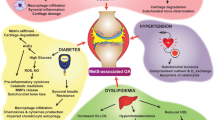
Obesity is a strong risk factor for incident of OA. The obesity-induced OA may be due to high mechanical stresses applied on the tissues. So some weight-bearing joints, particularly the knee and hip, are easily suffering from OA as a result of increased joint loading [3, 4]. And patients with knee OA would experience the symptomatic relief after weight loss [5]. However, various cohort studies have demonstrated that obesity is also a risk factor for non-weight bearing joints OA, such as hands [6]. Since the mechanical factor can’t explain this phenomenon, more and more teams are paying their attention to an obesity-related systemic factor on the pathogenesis of OA. Nowadays, obesity was commonly considered as a low grade inflammatory state. Adipose tissue was a real endocrine organ that releases cytokines, such as interleukin-1 (IL-1) and tumor necrosis factor-α (TNF-α), as well as adipokines, such as leptin, adiponectin, resistin, visfatin and so on [7]. The novel adipokine family exhibited pleiotropic functions through endocrine, paracrine, autocrine in a wide variety of physiological or physiopathological processes, including food take, energy expenditure, lipid and glucose metabolism, inflammation, insulin resistant, bone formation and so on [8, 9]. Adipokine was also viewed as a potential systemic factor which links obesity to arthritis [10]. This review addresses recent studies concerning the involvement of adipokines in OA, concentrating on the roles of adipokines played in the pathophysiology of OA.
Leptin
Leptin, a 16 kDa non-glycosylated protein secreted by adipose tissue, was encoded by the obese (ob) gene localized on 7 and 6 chromosomes in human and mouse. The levels of serum leptin were directly correlated with white adipose tissue (WAT) mass [11]. Leptin receptor (Ob-R) was a member in class I cytokine receptor superfamily and mediated various biological activity of leptin [12]. Structurally, both leptin and its receptor shared common structural and functional properties with IL-6, so this adipose-derived protein has been classified as an adipocytokine or adipokine [13]. By binding to Ob-R leptin triggered a signal cascade involving the JAK (janus kinase)/STAT (signal transducers and activators of transcription) family, as well as/PI3K/Akt/NF-κB and p300 signaling pathway [14, 15].
Leptin played a key role in the regulation of body weight by decreasing food intake and stimulating energy consumption [16]. The other function of leptin system has been widely studied and implicated as a regulatory molecule in various physiologic processes, such as infection, inflammation, autoimmune diseases, rheumatoid arthritis (RA) and so on [17–19].
Increasing evidence suggested that leptin was a novel proinflammatory adipocyte-derived factor in the pathophysiology of OA. Figenschau et al. demonstrated that both serially cultured human articular chondrocytes and native human cartilage expressed the Ob-R. And chondrocytes stimulated with leptin exhibited an increased proliferation and an enhanced synthesis of proteoglycans and collagen [20]. Another study by Dummond et al. found that leptin played a key role in the pathogenesis of OA. Their study was the first one to show the presence of leptin in synovial fluid (SF) obtained from OA patients and the significant correlation between leptin levels and BMI. Intra-articular injection of leptin strongly stimulated the synthesis of insulin-like growth factor-1 (IGF-1) and transforming growth factor-β (TGF-β) at both the messenger RNA (mRNA) and protein levels which might exert anabolic activity in cartilage metabolism [21]. Moreover, exogenous leptin could enhance chondrocytes proliferation and subsequent cell differentiation [22]. Actually, up-regulation the number of chondrocytes by leptin was a considerably effective way in the treatment of OA-affect articular cartilage. The process which increases the amount of chondrocytes in damaged cartilage by autologous chondrocyte transplantation could repair the chondral defects [23, 24]. It seemed that they all regarded leptin as an anabolic role in cartilage.
On the contrary, not all evidence supported the view above. Simopoulou and Ku et al. showed that SF leptin level significantly increased in advanced OA cartilage compared to minimal. The evidence that leptin induced IL-1β production and matrix metalloproteinases-9 (MMP-9), MMP-13 protein expression, indicated that leptin may act as a proinflammatory role on cartilage metabolism [25, 26]. Vuolteenaho et al. also found that leptin could mediate the cartilage metabolism by enhancing the production of NO, Prostaglandin E2 (PGE2), IL-6, and IL-8 in OA cartilage [27]. Using small interference RNA to induce leptin down-regulation could directly inhibit MMP-13 expression in chondrocytes [28]. Furthermore, treatment of ATDC5 chondrocytes and human primary chondrocytes with leptin pointed out that leptin played a proinflammatory role by inducing NO production and increasing the expression of nitric oxide synthase (NOS) type II in synergy with IL-1 [29]. All of these work focused on the proinflammatory effect in vitro and leptin seemed to a foe of cartilage homeostasis. Interesting, by comparing incidence of knee OA between leptin-deficient (ob/ob) and leptin receptor-deficient (db/db) female obese mice and controls, researcher failed to detect any significant between them. This novel-designed experiment suggested that leptin was a necessary role in the pathophysiology of the OA associated with obesity. Obesity alone was insufficient to induce systemic inflammation and knee OA [30].
Recently, our team has studied the role of leptin on normal articular cartilage in vivo. By injecting into the knee joints of rats with recombinant rat leptin, we found that proteolytic enzymes, including MMPs, and cysteine proteases, were markedly increased at both gene and protein levels, while anabolic factor, likes basic fibroblast growth factor (bFGF), was decreased. In addition, the gene expression of ADAMT-4 (a disintegrin and metalloproteinase with thrombospondin motifs) and ADAMT-5, which were considered as the most efficient aggrecanases in the pathogenesis of OA, were markedly increased after leptin treatment in our study [31–33]. These evidences clearly indicated that leptin may act as a catabolic factor involved in the progression of osteoarthritis [34].
Besides, it was now evident that leptin was also expressed in osteoblasts, stromal cells, and disc cell in the musculoskeleton system and could modulate bone growth [35–39]. Mutabaruka et al. showed that subchondral osteoblasts in OA exhibited high levels of leptin compared to normal and this local leptin production could respond for abnormal osteoblast function [40]. Osteophytes, synovium and infrapatellar fat pad could also secrete generous leptin to synovial fluid in OA [41]. These observations collectively demonstrated that leptin was also partly involved in or responded for the pathological change of other joint tissues in the pathophysiology of OA.
In a word, leptin was a double-edged sword, inducing both synthesis and degradation of articular cartilage. To date, the mechanism of leptin in the development of arthritis was still unclear and should be further investigated.
Adiponectin
Adiponectin, also called acrp30 (adipocyte complement-related protein of 30 kDa), was a newly discovered hormone secreted by adipocytes [42]. It shared sequence homology with collagen VIII, X and complement factor C1q [42, 43] and presented in three molecular forms: trimer, hexamer, and a high molecular weight (HMW) species [44]. In human beings, the circulating levels of adiponectin were decreased in obese and diabetic states. The biological activity of adiponectin was mediated by specific receptors, AdipoR1 and AdipoR2. AdipoR1 was abundantly expressed in skeletal muscle, whereas AdipoR2 was most abundantly in the liver [45]. Previously, it has been reported that 5′-AMP-activated protein kinase (AMPK)/p38/IKKαβ, NF-κB and c-Jun N-terminal kinases (JNKs) signaling pathway were involved in the adiponectin-mediated pathological processes [46, 47].
There was emerging evidence that adiponectin may have wide range of effects in cardiovascular disease, type 2 diabetes, and metabolic syndrome [48–50]. Previous reports have demonstrated that adiponectin was particularly important for inflammation. In vitro study, cytokines such as IL-6 and TNF-α could inhibit adiponectin secretion in cultured adipocytes [51, 52]. However, a negatively correlation between plasma adiponectin level and plasma C-reactive protein (CRP) level indicated that adiponectin may exert an anti-inflammatory role by regulating CRP expression in adipose tissue [53]. So sometimes adiponectin could exert anti-inflammatory rather than proinflammatory activities.
A growing number of investigations have consistently demonstrated that adiponectin played a dual role in arthritis. Adiponectin has been described as a potent mechanistic link between obesity and RA since its plasma level was higher in RA patients compares to healthy controls [54]. And another study about synovial fibroblasts in vitro revealed that adiponectin could stimulate the production of IL-6 and pro-MMP-1, which were regarded as key mediators of destructive arthritis [55]. So, the intracellular regulation of adiponectin might contribute to proinflammatory and matrix-degrading. As to OA, a present study revealed that OA plasma exhibited significantly 100-fold increase of adiponectin (5.3 ± 1.3 μg/ml) as compared with OA SF (44 ± 17 ng/ml) [56]. Synovium and infrapatellar fat pad have been shown to be the main sources of adiponectin in the OA-affect joint [41]. These findings provided evidence for a specific local dysregulation of adiponectin in the arthritis joint space. They also suggested adiponectin may act as a protective role against OA by up-regulating tissue inhibitor of metalloproteinase-2 (TIMP-2) and down-regulating IL-1β-induced MMP-13 at both mRNA and proteins levels [56]. In STR/Ort mice, a spontaneous primary osteoarthritis model, the serum adiponectin concentration was significantly low than in the control group [57]. All of these above puzzled us: did adiponectin play a protective role during the pathophysiology of OA?
Unfortunately, the role of adiponectin played in OA was controversial. Lago’s study demonstrated that adiponectin may have proinflammatory effects on chondrocytes by inducing the expression of NOS2 and stimulating proinflammatory cytokines release, such as IL-6, MMP-3, MMP-9 and monocyte chemattractant protein-1 (MCP-1) [58]. Surprisingly, Filkova et al. reported that increased serum levels of adiponectin in erosive OA was found compared with non-erosive OA, suggesting that adiponectin may play a role in matrix degradations [59]. Also, by stimulating the syntheses of vascular endothelial growth factor (VEGF) and MMPs, adiponectin facilitated joint inflammation and destruction [60]. All of the above reveal the close relationship between adiponectin and OA, no matter by protecting the joints or facilitating the osteoarthritis.
Resistin
Resistin, known as a macrophage/monocyte-derived proinflammatory mediator, belonged to the found in inflammatory zone (FIZZ) protein family and secreted mainly by peripheral-blood mononuclear cells [61–63]. Resistin received its name from its apparent induction of insulin resistance in mice [64]. It was secreted by adipose tissue, but also expressed in several other tissues such as neutrophils, lung, heart, and synovial tissue [62, 65]. In rodents, circulating levels of resistin were increased in obesity [66]. In recent years a growing number of studies proved that resistin has been implicated in inflammatory processes. It was reported that human resistin stimulated the synthesis and secretion of TNF-α and IL-12 and this involved the activation of NF-κB transcription factor in macrophages in vitro [67]. Moreover, proinflammatory cytokines including IL-1, IL-6, TNF-α, and lipopolysaccharides (LPS) could stimulate the up-expression of resistin mRNA [68].
Recent studies have shown that resistin made plenty of biological effects to arthritis. Resistin could be detected locally in the inflamed joints of patients with RA and OA and elevated in RA [41, 69]. It could induce arthritis when injected into healthy mouse joints and strongly up-regulated the release of proinflammatory cytokines such as TNF-α, IL-1β, and IL-6 [69]. Furthermore, markedly increased production of resistin was found at local sites of inflammation such as synovial tissue and synovial fluid in patients with rheumatoid arthritis [65]. To note, recombinant resistin stimulated proteoglycan degradation in mouse femoral head cultures and induction of inflammatory cytokine and PGE2 production, whereas it inhibited proteoglycan synthesis in human cartilage explants [70]. As noted above, it has been concluded that resistin may act as a novel proinflammatory mediator in chronic joint inflammatory diseases.
Visfatin
Visfatin was previously identified as a presumptive cytokine named pre-B cell colony-enhancing factor (PBEF) which synergized with IL-7 to promote the differentiation of B-cell precursors. It was coded by a novel gene which was isolated from a human peripheral blood lymphocyte cDNA library [71]. Several tissues expressed visfatin/PBEF, including skeletal muscle, liver, and bone marrow [71]. Visfatin was closely correlated with the regulation of insulin secretion, insulin receptor signaling as well as mRNA levels of diabetes-related genes in mouse [72, 73]. It was also up-regulated in acute lung injury, sepsis and atherosclerotic lesions [74, 75], while it was shown to induce the production of IL-1β, IL-6, IL-10, and TNF-α in human monocytes in vitro [76]. In turn, TNF-α, IL-1β, IL-6, LPS and dexamethasone could stimulate the synthesis of visfatin. All of above suggested that visfatin should be regarded as an inflammatory mediator in some circumstances.
There were indications that visfatin may also be involved in the pathogenesis of RA and OA. The expression of visfatin was detected in synovial tissue, serum, and synovial fluid and a marked higher level of serum visfatin was noted in patients with RA [54, 77]. Levels of visfatin in serum and synovial fluid were strong positive correlated with the severity of RA. Moreover, visfatin presented a proinflammatory and matrix-degrading role by inducing IL-6, IL-8, MMP-1, and MMP-3 production in RA synovial fibroblasts [77]. Nowell and colleagues demonstrated that visfatin was regulated via IL-6 trans-signaling and the IL-6-related cytokine oncostatin M [78]. Recently, a study focused on the role of visfatin in OA indicated that visfatin synthesis was increased by IL-1β treatment in vitro culture of human chondrocytes. Meanwhile, visfatin manifested a pro-degradative effect by increasing the synthesis and release of MMP-3, MMP-13, ADAMTS-4, and ADAMTS-5 and decreasing aggrecan production in chondrocyte, suggesting that visfatin had a catabolic function in cartilage and might have an important role in the pathophysiology of OA [79].
Adipokine, obesity and osteoarthritis: more complex than predicted
Given all this evidence, studies on different adipokines revealed that they have played proinflammatory and catabolic/anabolic role during the pathophysiology of OA. Recently, two newly discovered adipokines named vaspin and omentin, have been detected in synovial fluid obtained from RA and OA patients [80]. So the relationship between obesity and osteoarthritis was more complex than predicted!
In conclusion, adipokines exert lots of biological effects to joint and contribute to the progress of OA. Nevertheless, OA is a multifactorial disease and the exact mechanism of adipokines in obesity-induced OA is still unclear. Further investigations are needed to clarify their roles in OA.
References
Felson DT, Chaisson CE, Hill CL, Totterman SM, Gale ME, Skinner KM, Kazis L, Gale DR (2001) The association of bone marrow lesions with pain in knee osteoarthritis. Ann Intern Med 134:541–549
Pottie P, Presle N, Terlain B, Netter P, Mainard D, Berenbaum F (2006) Obesity and osteoarthritis: more complex than predicted!. Ann Rheum Dis 65:1403–1405
Felson DT, Anderson JJ, Naimark A, Walker AM, Meenan RF (1988) Obesity and knee osteoarthritis. The Framingham Study. Ann Intern Med 109:18–24
Oliveria SA, Felson DT, Cirillo PA, Reed JI, Walker AM (1999) Body weight, body mass index, and incident symptomatic osteoarthritis of the hand, hip, and knee. Epidemiology 10:161–166
Christensen R, Bartels EM, Astrup A, Bliddal H (2007) Effect of weight reduction in obese patients diagnosed with knee osteoarthritis: a systematic review and meta-analysis. Ann Rheum Dis 66:433–439
Carman WJ, Sowers M, Hawthorne VM, Weissfeld LA (1994) Obesity as a risk factor for osteoarthritis of the hand and wrist: a prospective study. Am J Epidemiol 139:119–129
Kershaw EE, Flier JS (2004) Adipose tissue as an endocrine organ. J Clin Endocrinol Metab 89:2548–2556
Rabe K, Lehrke M, Parhofer KG, Broedl UC (2008) Adipokines and insulin resistance. Mol Med 14:741–751
Gualillo O, Gonzalez-Juanatey JR, Lago F (2007) The emerging role of adipokines as mediators of cardiovascular function: physiologic and clinical perspectives. Trends Cardiovasc Med 17:275–283
Gabay O, Berenbaum F (2009) Adipokines in Arthritis: new kids on the block. Curr Rheumatol Rev 5:226–232
Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM (1994) Positional cloning of the mouse obese gene and its human homologue. Nature 372:425–432
Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J, Muir C, Sanker S, Moriarty A, Moore KJ, Smutko JS, Mays GG, Wool EA, Monroe CA, Tepper RI (1995) Identification and expression cloning of a leptin receptor, OB-R. Cell 83:1263–1271
Baumann H, Morella KK, White DW, Dembski M, Bailon PS, Kim H, Lai CF, Tartaglia LA (1996) The full-length leptin receptor has signaling capabilities of interleukin 6-type cytokine receptors. Proc Natl Acad Sci USA 93:8374–8378
Tong KM, Shieh DC, Chen CP, Tzeng CY, Wang SP, Huang KC, Chiu YC, Fong YC, Tang CH (2008) Leptin induces IL-8 expression via leptin receptor, IRS-1, PI3 K, Akt cascade and promotion of NF-kappaB/p300 binding in human synovial fibroblasts. Cell Signal 20:1478–1488
Tang CH, Lu DY, Yang RS, Tsai HY, Kao MC, Fu WM, Chen YF (2007) Leptin-induced IL-6 production is mediated by leptin receptor, insulin receptor substrate-1, phosphatidylinositol 3-kinase, Akt, NF-kappaB, and p300 pathway in microglia. J Immunol 179:1292–1302
Friedman JM, Halaas JL (1998) Leptin and the regulation of body weight in mammals. Nature 395:763–770
Faggioni R, Feingold KR, Grunfeld C (2001) Leptin regulation of the immune response and the immunodeficiency of malnutrition. FASEB J 15:2565–2571
Siegmund B, Sennello JA, Jones-Carson J, Gamboni-Robertson F, Lehr HA, Batra A, Fedke I, Zeitz M, Fantuzzi G (2004) Leptin receptor expression on T lymphocytes modulates chronic intestinal inflammation in mice. Gut 53:965–972
Bokarewa M, Bokarew D, Hultgren O, Tarkowski A (2003) Leptin consumption in the inflamed joints of patients with rheumatoid arthritis. Ann Rheum Dis 62:952–956
Figenschau Y, Knutsen G, Shahazeydi S, Johansen O, Sveinbjornsson B (2001) Human articular chondrocytes express functional leptin receptors. Biochem Biophys Res Commun 287:190–197
Dumond H, Presle N, Terlain B, Mainard D, Loeuille D, Netter P, Pottie P (2003) Evidence for a key role of leptin in osteoarthritis. Arthritis Rheum 48:3118–3129
Nakajima R, Inada H, Koike T, Yamano T (2003) Effects of leptin to cultured growth plate chondrocytes. Horm Res 60:91–98
Blanco FJ, Guitian R, Vazquez-Martul E, de Toro FJ, Galdo F (1998) Osteoarthritis chondrocytes die by apoptosis A possible pathway for osteoarthritis pathology. Arthritis Rheum 41:284–289
Brittberg M, Lindahl A, Nilsson A, Ohlsson C, Isaksson O, Peterson L (1994) Treatment of deep cartilage defects in the knee with autologous chondrocyte transplantation. N Engl J Med 331:889–895
Simopoulou T, Malizos KN, Iliopoulos D, Stefanou N, Papatheodorou L, Ioannou M, Tsezou A (2007) Differential expression of leptin and leptin’s receptor isoform (Ob-Rb) mRNA between advanced and minimally affected osteoarthritic cartilage; effect on cartilage metabolism. Osteoarthritis Cartilage 15:872–883
Ku JH, Lee CK, Joo BS, An BM, Choi SH, Wang TH, Cho HL (2009) Correlation of synovial fluid leptin concentrations with the severity of osteoarthritis. Clin Rheumatol 28:1431–1435
Vuolteenaho K, Koskinen A, Kukkonen M, Nieminen R, Paivarinta U, Moilanen T, Moilanen E (2009) Leptin enhances synthesis of proinflammatory mediators in human osteoarthritic cartilage-mediator role of NO in leptin-induced PGE2, IL-6, and IL-8 production. Mediators Inflamm 2009:345838
Iliopoulos D, Malizos KN, Tsezou A (2007) Epigenetic regulation of leptin affects MMP-13 expression in osteoarthritic chondrocytes: possible molecular target for osteoarthritis therapeutic intervention. Ann Rheum Dis 66:1616–1621
Otero M, Lago R, Lago F, Reino JJ, Gualillo O (2005) Signalling pathway involved in nitric oxide synthase type II activation in chondrocytes: synergistic effect of leptin with interleukin-1. Arthritis Res Ther 7:R581–R591
Griffin TM, Huebner JL, Kraus VB, Guilak F (2009) Extreme obesity due to impaired leptin signaling in mice does not cause knee osteoarthritis. Arthritis Rheum 60:2935–2944
Thirunavukkarasu K, Pei Y, Wei T (2007) Characterization of the human ADAMTS-5 (aggrecanase-2) gene promoter. Mol Biol Rep 34:225–231
Malfait AM, Liu RQ, Ijiri K, Komiya S, Tortorella MD (2002) Inhibition of ADAM-TS4 and ADAM-TS5 prevents aggrecan degradation in osteoarthritic cartilage. J Biol Chem 277:22201–22208
Huang K, Wu LD (2010) Suppression of aggrecanase: a novel protective mechanism of dehydroepiandrosterone in osteoarthritis? Mol Biol Rep 37:1241–1245
Bao JP, Chen WP, Feng J, Hu PF, Shi ZL, Wu LD (2009) Leptin plays a catabolic role on articular cartilage. Mol Biol Rep. doi:10.1007/s11033-009-9911-x
Morroni M, De Matteis R, Palumbo C, Ferretti M, Villa I, Rubinacci A, Cinti S, Marotti G (2004) In vivo leptin expression in cartilage and bone cells of growing rats and adult humans. J Anat 205:291–296
Reseland JE, Syversen U, Bakke I, Qvigstad G, Eide LG, Hjertner O, Gordeladze JO, Drevon CA (2001) Leptin is expressed in and secreted from primary cultures of human osteoblasts and promotes bone mineralization. J Bone Miner Res 16:1426–1433
Zhao CQ, Liu D, Li H, Jiang LS, Dai LY (2008) Expression of leptin and its functional receptor on disc cells: contribution to cell proliferation. Spine (Phila Pa 1976) 33:E858–864
Lee YJ, Park JH, Ju SK, You KH, Ko JS, Kim HM (2002) Leptin receptor isoform expression in rat osteoblasts and their functional analysis. FEBS Lett 528:43–47
Reseland JE, Gordeladze JO (2002) Role of leptin in bone growth: central player or peripheral supporter? FEBS Lett 528:40–42
Mutabaruka MS, Aoulad Aissa M, Delalandre A, Lavigne M, Lajeunesse D (2010) Local leptin production in osteoarthritis subchondral osteoblasts may be responsible for their abnormal phenotypic expression. Arthritis Res Ther 12:R20
Presle N, Pottie P, Dumond H, Guillaume C, Lapicque F, Pallu S, Mainard D, Netter P, Terlain B (2006) Differential distribution of adipokines between serum and synovial fluid in patients with osteoarthritis. Contribution of joint tissues to their articular production. Osteoarthritis Cartilage 14:690–695
Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF (1995) A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem 270:26746–26749
Hu E, Liang P, Spiegelman BM (1996) AdipoQ is a novel adipose-specific gene dysregulated in obesity. J Biol Chem 271:10697–10703
Pajvani UB, Du X, Combs TP, Berg AH, Rajala MW, Schulthess T, Engel J, Brownlee M, Scherer PE (2003) Structure-function studies of the adipocyte-secreted hormone Acrp30/adiponectin. Implications fpr metabolic regulation and bioactivity. J Biol Chem 278:9073–9085
Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, Murakami K, Ohteki T, Uchida S, Takekawa S, Waki H, Tsuno NH, Shibata Y, Terauchi Y, Froguel P, Tobe K, Koyasu S, Taira K, Kitamura T, Shimizu T, Nagai R, Kadowaki T (2003) Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 423:762–769
Tang CH, Chiu YC, Tan TW, Yang RS, Fu WM (2007) Adiponectin enhances IL-6 production in human synovial fibroblast via an AdipoR1 receptor, AMPK, p38, and NF-kappa B pathway. J Immunol 179:5483–5492
Tilg H, Kaser A (2009) Adiponectin and JNK: metabolic/inflammatory pathways affecting gastrointestinal carcinogenesis. Gut 58:1576–1577
Weyer C, Funahashi T, Tanaka S, Hotta K, Matsuzawa Y, Pratley RE, Tataranni PA (2001) Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab 86:1930–1935
Funahashi T, Nakamura T, Shimomura I, Maeda K, Kuriyama H, Takahashi M, Arita Y, Kihara S, Matsuzawa Y (1999) Role of adipocytokines on the pathogenesis of atherosclerosis in visceral obesity. Intern Med 38:202–206
Gilardini L, McTernan PG, Girola A, da Silva NF, Alberti L, Kumar S, Invitti C (2006) Adiponectin is a candidate marker of metabolic syndrome in obese children and adolescents. Atherosclerosis 189:401–407
Bruun JM, Lihn AS, Verdich C, Pedersen SB, Toubro S, Astrup A, Richelsen B (2003) Regulation of adiponectin by adipose tissue-derived cytokines: in vivo and in vitro investigations in humans. Am J Physiol Endocrinol Metab 285:E527–E533
Fasshauer M, Kralisch S, Klier M, Lossner U, Bluher M, Klein J, Paschke R (2003) Adiponectin gene expression and secretion is inhibited by interleukin-6 in 3T3-L1 adipocytes. Biochem Biophys Res Commun 301:1045–1050
Engeli S, Feldpausch M, Gorzelniak K, Hartwig F, Heintze U, Janke J, Mohlig M, Pfeiffer AF, Luft FC, Sharma AM (2003) Association between adiponectin and mediators of inflammation in obese women. Diabetes 52:942–947
Otero M, Lago R, Gomez R, Lago F, Dieguez C, Gomez-Reino JJ, Gualillo O (2006) Changes in plasma levels of fat-derived hormones adiponectin, leptin, resistin and visfatin in patients with rheumatoid arthritis. Ann Rheum Dis 65:1198–1201
Ehling A, Schaffler A, Herfarth H, Tarner IH, Anders S, Distler O, Paul G, Distler J, Gay S, Scholmerich J, Neumann E, Muller-Ladner U (2006) The potential of adiponectin in driving arthritis. J Immunol 176:4468–4478
Chen TH, Chen L, Hsieh MS, Chang CP, Chou DT, Tsai SH (2006) Evidence for a protective role for adiponectin in osteoarthritis. Biochim Biophys Acta 1762:711–718
Uchida K, Urabe K, Naruse K, Ogawa Z, Mabuchi K, Itoman M (2009) Hyperlipidemia and hyperinsulinemia in the spontaneous osteoarthritis mouse model, STR/Ort. Exp Anim 58:181–187
Lago R, Gomez R, Otero M, Lago F, Gallego R, Dieguez C, Gomez-Reino JJ, Gualillo O (2008) A new player in cartilage homeostasis: adiponectin induces nitric oxide synthase type II and pro-inflammatory cytokines in chondrocytes. Osteoarthritis Cartilage 16:1101–1109
Filkova M, Liskova M, Hulejova H, Haluzik M, Gatterova J, Pavelkova A, Pavelka K, Gay S, Muller-Ladner U, Senolt L (2009) Increased serum adiponectin levels in female patients with erosive compared with non-erosive osteoarthritis. Ann Rheum Dis 68:295–296
Choi HM, Lee YA, Lee SH, Hong SJ, Hahm DH, Choi SY, Yang HI, Yoo MC, Kim KS (2009) Adiponectin may contribute to synovitis and joint destruction in rheumatoid arthritis by stimulating vascular endothelial growth factor, matrix metalloproteinase-1, and matrix metalloproteinase-13 expression in fibroblast-like synoviocytes more than proinflammatory mediators. Arthritis Res Ther 11:R161
Steppan CM, Brown EJ, Wright CM, Bhat S, Banerjee RR, Dai CY, Enders GH, Silberg DG, Wen X, Wu GD, Lazar MA (2001) A family of tissue-specific resistin-like molecules. Proc Natl Acad Sci USA 98:502–506
Patel L, Buckels AC, Kinghorn IJ, Murdock PR, Holbrook JD, Plumpton C, Macphee CH, Smith SA (2003) Resistin is expressed in human macrophages and directly regulated by PPAR gamma activators. Biochem Biophys Res Commun 300:472–476
Savage DB, Sewter CP, Klenk ES, Segal DG, Vidal-Puig A, Considine RV, O’Rahilly S (2001) Resistin/Fizz3 expression in relation to obesity and peroxisome proliferator-activated receptor-gamma action in humans. Diabetes 50:2199–2202
Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, Patel HR, Ahima RS, Lazar MA (2001) The hormone resistin links obesity to diabetes. Nature 409:307–312
Senolt L, Housa D, Vernerova Z, Jirasek T, Svobodova R, Veigl D, Anderlova K, Muller-Ladner U, Pavelka K, Haluzik M (2007) Resistin in rheumatoid arthritis synovial tissue, synovial fluid and serum. Ann Rheum Dis 66:458–463
Rajala MW, Qi Y, Patel HR, Takahashi N, Banerjee R, Pajvani UB, Sinha MK, Gingerich RL, Scherer PE, Ahima RS (2004) Regulation of resistin expression and circulating levels in obesity, diabetes, and fasting. Diabetes 53:1671–1679
Silswal N, Singh AK, Aruna B, Mukhopadhyay S, Ghosh S, Ehtesham NZ (2005) Human resistin stimulates the pro-inflammatory cytokines TNF-alpha and IL-12 in macrophages by NF-kappaB-dependent pathway. Biochem Biophys Res Commun 334:1092–1101
Kaser S, Kaser A, Sandhofer A, Ebenbichler CF, Tilg H, Patsch JR (2003) Resistin messenger-RNA expression is increased by proinflammatory cytokines in vitro. Biochem Biophys Res Commun 309:286–290
Bokarewa M, Nagaev I, Dahlberg L, Smith U, Tarkowski A (2005) Resistin, an adipokine with potent proinflammatory properties. J Immunol 174:5789–5795
Lee JH, Ort T, Ma K, Picha K, Carton J, Marsters PA, Lohmander LS, Baribaud F, Song XY, Blake S (2009) Resistin is elevated following traumatic joint injury and causes matrix degradation and release of inflammatory cytokines from articular cartilage in vitro. Osteoarthritis Cartilage 17:613–620
Samal B, Sun Y, Stearns G, Xie C, Suggs S, McNiece I (1994) Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol Cell Biol 14:1431–1437
Brown JE, Onyango DJ, Ramanjaneya M, Conner AC, Patel ST, Dunmore SJ, Randeva HS (2010) Visfatin regulates insulin secretion, insulin receptor signalling and mRNA expression of diabetes-related genes in mouse pancreatic beta-cells. J Mol Endocrinol 44:171–178
Revollo JR, Korner A, Mills KF, Satoh A, Wang T, Garten A, Dasgupta B, Sasaki Y, Wolberger C, Townsend RR, Milbrandt J, Kiess W, Imai S (2007) Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab 6:363–375
Dahl TB, Yndestad A, Skjelland M, Oie E, Dahl A, Michelsen A, Damas JK, Tunheim SH, Ueland T, Smith C, Bendz B, Tonstad S, Gullestad L, Froland SS, Krohg-Sorensen K, Russell D, Aukrust P, Halvorsen B (2007) Increased expression of visfatin in macrophages of human unstable carotid and coronary atherosclerosis: possible role in inflammation and plaque destabilization. Circulation 115:972–980
Jia SH, Li Y, Parodo J, Kapus A, Fan L, Rotstein OD, Marshall JC (2004) Pre-B cell colony-enhancing factor inhibits neutrophil apoptosis in experimental inflammation and clinical sepsis. J Clin Invest 113:1318–1327
Moschen AR, Kaser A, Enrich B, Mosheimer B, Theurl M, Niederegger H, Tilg H (2007) Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J Immunol 178:1748–1758
Brentano F, Schorr O, Ospelt C, Stanczyk J, Gay RE, Gay S, Kyburz D (2007) Pre-B cell colony-enhancing factor/visfatin, a new marker of inflammation in rheumatoid arthritis with proinflammatory and matrix-degrading activities. Arthritis Rheum 56:2829–2839
Nowell MA, Richards PJ, Fielding CA, Ognjanovic S, Topley N, Williams AS, Bryant-Greenwood G, Jones SA (2006) Regulation of pre-B cell colony-enhancing factor by STAT-3-dependent interleukin-6 trans-signaling: implications in the pathogenesis of rheumatoid arthritis. Arthritis Rheum 54:2084–2095
Gosset M, Berenbaum F, Salvat C, Sautet A, Pigenet A, Tahiri K, Jacques C (2008) Crucial role of visfatin/pre-B cell colony-enhancing factor in matrix degradation and prostaglandin E2 synthesis in chondrocytes: possible influence on osteoarthritis. Arthritis Rheum 58:1399–1409
Senolt L, Polanska M, Filkova M, Oslejskova L, Pavelka K, Gay S, Haluzik M, Vencovsky J (2009) Vaspin and omentin: new adipokines differentially regulated at the site of inflammation in rheumatoid arthritis. Ann Rheum Dis. doi:10.1136/ard.2009.119735
Acknowledgment
This work was supported by grants from Health Bureau of Zhejiang Province (2006A055).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hu, Pf., Bao, Jp. & Wu, Ld. The emerging role of adipokines in osteoarthritis: a narrative review. Mol Biol Rep 38, 873–878 (2011). https://doi.org/10.1007/s11033-010-0179-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-010-0179-y