
Abstract
Brassinosteroids (BRs) are a class of plant-specific steroid hormones and play an essential role in plant growth and development. The DWARF4 (DWF4) gene encodes a C-22 hydroxylase, which is a rate-limiting enzyme in BR biosynthesis pathway. Here, the DWF4 (ZmDWF4) in maize (Zea mays L.), the ortholog of Arabidopsis DWF4 (CYP90B1), was transformed into elite inbred Q319 and driven maize ubiquitin promoter. In transgenic lines, the level of the endogenous BR precursor canosterone (CS) was significantly enhanced. ZmDWF4 overexpression greatly improved grain yield per ear from 28.3 to 33.5% in transgenic lines compared with that in non-transgenic Q319 plants. Moreover, ZmDWF4 overexpression significantly enhanced heterosis of combinations by increasing both seed number and seed weight up to 20.4% in transgenic hybrids. Further analysis revealed that ZmDWF4 overexpression significantly improved several agronomic traits and leaf photosynthetic ability in transgenic lines. Finally, RNA-seq analysis suggested that overexpressing ZmDWF4 improved cell growth, cell division, and nutrient assimilation in transgenic kernels. Altogether, our results demonstrate that manipulating BR level by overexpressing ZmDWF4 effectively improves maize agronomic traits, and thus, provides a useful biotechnological strategy for the genetic improvement and further commercially favorable maize breeding.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Maize is one of the most important crops in the world. Using transgenic engineering to genetically improve yield traits has been considered as an effective strategy to alleviate the increasingly prominent conflicts between crop supply and global food demands. In recent years, functional genes have been chosen as the targets of genetic manipulation for improving grain yields in terms of promoting starch synthesis, cell division, and sugar transport (Hannah et al. 2012; Li et al. 2013; Sun et al. 2017; Wu et al. 2008; Wu et al. 2019; Xie et al. 2018).
Brassinosteroids (BRs) are plant-specific steroid hormones that play important roles in plant growth and development by regulating cell elongation, division, and differentiation (Fridman and Savaldi-Goldstein 2013; Singh and Savaldi-Goldstein 2015; Tian et al. 2018; Tong et al. 2014; Xia et al. 2015). The study demonstrate that BRs are essential for various plant development processes, such as pollen development, root growth, flowering time, and seed production (Best et al. 2016; Choudhary et al. 2012; Feng et al. 2016; Kir et al. 2015; Liu et al. 2016; Shimada et al. 2015; Singh et al. 2016; Tong et al. 2012; Vogler et al. 2014; Wei and Li 2016; Zhang et al. 2014a; Zhang et al. 2012). The classical BR-deficient or BR-insensitive mutants show diverse phenotypes, including dwarf plant architecture, small leaf blade, dark green leaves, abnormal vascular tissue, delayed flowering, delayed aging, and reduced fertility (Guo et al. 2013; Li et al. 1996; Szekeres et al. 1996; Wang et al. 2012).
The BR biosynthesis process has been well characterized. Campesterol was converted into campestanol, then followed by the formation of canosterone (CS), eventually leading to the formation of brassinolide (BL) from CS (Fujioka et al. 1998). Notably, several key genes involved in BR biosynthesis have been identified in Arabidopsis, for example, de-etiolated-2 (DET2) encoding a steroid 5α-reductase, DWF4 encoding a C-22 hydroxylase, and constitutive photomorphogenesis and dwarfism (CPD) encoding a C-23 hydroxylase (Choe et al. 1998; Fujioka et al. 1998; Li et al. 1996; Szekeres et al. 1996). Moreover, several key BR biosynthetic genes have been identified in maize. A classical dwarf maize plant nana plant1 (na1), was characterized, and NA1 is an ortholog of Arabidopsis DET2 that catalyzes the limited step of BR synthesis (Hartwig et al. 2011). The NA2 gene, which is involved in plant height, tillers, leaf morphology, and tassel development, encodes a maize ortholog of the Arabidopsis BR biosynthesis gene DWF1 (Best et al. 2016).
DWF4 belongs to P450 family and functions as the key rate-limiting enzyme in a BR biosynthesis pathway (Choe et al. 1998; Fujita et al. 2006). In Arabidopsis, overexpression of ZmDWF4 gene, has increased plant height, branch number, and seed yield (Liu et al. 2007), whereas overexpression of the Populus euphratica PeDWF4 gene in Arabidopsis has reduced seed yield and delayed flowering time (Si et al. 2016). These results suggested that DWF4 and its orthologs have diverse functions in different plant species. Here, we introduced ZmDWF4 driven by a maize ubiquitin promoter into maize plants. Independent ZmDWF4-overexpressing (ZmDWF4-OE) transgenic maize lines and their hybrid combinations showed enhanced BR level and significantly improved grain yields including increased seed number and grain weight. Furthermore, we showed that the overexpression of ZmDWF4 resulted in enlarged leaf area, increased photosynthesis, delayed leaf senescence, and significantly changed the transcription factors involved in grain filling. This study provides an alternative for genetic breeding through biotechnology in maize.
Materials and methods
Plant materials
Maize (Zea mays L.) inbred Q319 was grown in the field at the Experimental Station of Shandong Agricultural University (Tai’an, Shandong, China) and used as the transgenic receptor. T0 transgenic plants were transferred from the growth chamber to the greenhouse, and T1 to T4 transgenic lines were planted at the Experimental Station. T1 to T4 transgenic lines were generated by self-pollination.
For hybrid combinations, the T4 plants of the ZmDWF4-OE transgenic lines L100, L166, and L225 were crossed as the maternal parents with different inbred lines, including 9801, 3841, V3B1, and Zheng58 (Z58). Non-transgenic Q319 was crossed with these four inbred lines, and their F1 hybrids were considered as wild-type controls.
ZmDWF4 gene cloning and overexpressing vector construction
The full-length ZmDWF4 (Zm00001d028325) cDNA was amplified by PCR using RNA from two-week old seedling leaves of the maize inbred Q319 with the primer pair ZmDWF4-F and ZmDWF4-R. All primers used in this study are listed in Table S3. After verification by DNA sequencing, ZmDWF4 cDNA was inserted into pPZP211 vector downstream of maize ubiquitin promoter (Ubipro) with restriction enzyme sites BamHI/KpnI. The recombinant plasmid was designed as pPZP211-Ubipro::ZmDWF4 and was introduced into Agrobacterium tumefaciens LBA4404 for subsequent maize transformation.
Maize transformation and characterization of transgenic maize plants
To obtain ZmDWF4-overexpressing transgenic maize, the Agrobacterium-mediated genetic transformation was carried out as described by Ishida et al. (2007)). Briefly, immature embryos (11–13 days after pollination, DAP) were isolated and infected by Agrobacterium tumefaciens LBA4404 with the plasmid pPZP211-Ubipro::ZmDWF4. Kanamycin-resistant maize seedlings were transplanted in the greenhouse and were verified by PCR. Total genomic DNA was isolated from young leaves of the T0 transgenic maize plants and wild-type controls using the 2 × CTAB (hexadecyl trimethyl ammonium bromide) buffer, and then amplified using 2 × Taq Plus Master Mix (Dye Plus) (Vazyme Biotech, China). The PCR was performed with the following procedures: 94 °C for 5 min; 35 cycles of 94 °C for 30 s, 56 °C for 30 s, 72 °C for 1 min; 72 °C for 5 min. T1 to T4 transgenic progenies were identified by both PCR analysis and obtained by self-pollination. To distinguish the transgene with the endogenous gene, a forward primer named US5-1 was designed at the 3′ end of maize ubiquitin promoter and the reverse primer named ZmDWF4-A was designed at the coding region of ZmDWF4 gene.
Southern blotting analysis to confirm transgenic plant
Genomic DNA was extracted from young leaves of transgenic T4 generation maize plants using the 2× CTAB buffer as described by Zhao et al. (2016). Forty-five micrograms of genomic DNA was digested with BamHI and further separated with 0.8% agarose gel, and then the DNA was transferred onto nylon membranes (GE Healthcare, Mannheim, Germany). The PCR product amplified by the primer pair ZmDWF4SF and ZmDWF4SR was labeled with digoxin-dUTP (Roche, Mannheim, Germany). Southern hybridization was performed with DIG High Prime Labeling and Detection Starter Kit I as described in the manufacturer’s protocol (Roche). The hybridization signals on the nylon membrane were detected with a ChemiDOC™ XRS+ (BIO-RAD, Hercules, America).
Western blotting
Total proteins were extracted from young leaves as described by Ren et al. (2020). The proteins were separated using SDS-PAGE and then transferred onto a polyvinylidene difluoride (PVDF) membrane (Sigma-Aldrich, Merck-Millipore, Darmstadt, Germany). The membranes were blocked using 5% skim milk for 2 h at room temperature. Then, the membrane was incubated with primary antibodies for 2 h and secondary antibodies for 1 h at room temperature. Secondary antibodies were visualized using Pierce™ ECL Western Blotting Substrate (Thermo Fisher Scientific). The recombinant ZmDWF4 protein was used as the antigen, and the antibodies were prepared in rabbits by Abclonal (Abclonal Technology, Wuhan, China). The dilution of antibodies against ZmDWF4 was 1:1000. Antibodies against actin were obtained from Agrisera (www.agrisera.com) and were used according to the instructions.
qPCR analysis
Total RNA was extracted from leaves, roots, 5 days after pollination (DAP), 10 DAP, and 15 DAP kernels as described by Li et al. (2015), respectively. FastQuant RT Kit (TIANGEN BIOTECH, Beijing, China) was used to synthesize the first-stand cDNA. Expression of genes was analyzed by quantitative real-time PCR (qPCR) using SuperReal PreMix Plus (TIANGEN BIOTECH, Beijing, China) on the Bio-Rad CFX96 quantitative PCR system (Bio-Rad Laboratories, Inc., USA). The primer pairs for qPCR are listed in Table S3. The UBIQUITIN gene was used as an internal reference to normalize all data. The relative expression fold changes were calculated by the 2−ΔΔCt method.
Endogenous BR content determination analysis
The T4 ZmDWF4-OE transgenic line L100 and wild-type inbred were planted in the greenhouse. We sampled ear leaves from L100 and wild-type plants at the anthesis stage. BRs were measured in the phytohormone platform of the Institute of Genetics and Developmental Biology, Chinese Academy of Sciences according to the Xin’s method (Xin et al. 2018). Three biological repeats were ultimately used to assay endogenous BRs level.
Yield trials
For comparing the grain yield traits of T4 ZmDWF4-OE transgenic inbred lines and Q319 wild-type plants, field trials were carried out in Tai’an in 2015. Each line was planted in four-row plots with a row spacing of 65 cm and plant spacing of 25 cm in three replicates. Ten ears of each line were measured for ear length, ear diameter, kernel row number, and grain number per row. Kernel traits were measured when the seeds were dried till constant weight in 42 °C; the traits measured were 1000-grain weight, kernel length, kernel width, kernel thickness with three replicates of ten seeds of each line.
Field trials for comparing the yield parameters of ZmDWF4-OE hybrid combinations were carried out in Tai’an in 2016. The F1 hybrid plants were planted at 67500 plants per hectare on two-row plots of three replicates in the field. At 10 DAP, agronomic traits of the plants were measured, including plant height, ear height, leaf number, and leaf area. After harvest, ear traits and kernel traits were measured using the same method as above. Ten plants of each hybrid combination were randomly selected for measuring ear length, kernel length, kernel width, kernel thickness, and grain weight.
Phenotype analysis of transgenic lines
Seeds of T4 ZmDWF4-OE transgenic lines L100, L166, and L225, as well as the wild-type control (inbred Q319), were planted in the greenhouse. Plant traits were measured at the six-leaf stage, including plant height and leaf areas (from bottom to top, the fourth, fifth, and sixth leaves) with ten replicates of each transgenic line. At 10 DAP, we recorded agronomic traits of transgenic lines, including leaf number, photosynthesis rate, and areas of upper- and lower-ear leaves (UEL and LEL) and the ear leaf (EL). Ten plants of each transgenic line were sampled to measure the photosynthesis rate using the CIRAS-2 photosynthetic instrument (PP-systems, Hitchin, Hertfordshire, UK). Chlorophyll indices of ear leaves (15 cm from the leaf sheath) were measured at 0 DAP, 10 DAP, 20 DAP, 30 DAP, and 40 DAP using the CCM-200 chlorophyll content index tester (Zhirun, Beijing, China) as parameters to assess leaf senescence.
RNA-seq analysis
Total RNA was extracted from the seeds (10 and 15 DAP) of the ZmDWF4-OE transgenic line L100 and wild-type plants. The cDNA libraries were constructed in BIOMARKER, and original reads were sequenced based on an Illumina HiSeq2000 (Illumina, Inc., USA), and by rRNA and low-quality-fragment filtering, we obtained 99.30 Gb of high-quality, cleaned reads. The RNA-seq data was analyzed as described by Zhang et al. (2014b). GO (Gene Ontology) enrichment of differentially expression genes (DEGs) were analyzed in Metascape Enrichment Analysis tool (http://metascape.org/). Gene annotations were based on the AGPv3 5b60 gene annotation system (https://www.maizegdb.org/). Gene function classifications were based on the Omicshare tool (http://www.omicshare.com/).
We have submitted maize seed RNA-seq raw data (GSE108628, http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE108628) to the NCBI GEO database.
Results
Generation of transgenic maize with ZmDWF4 overexpression
To determine the role of ZmDWF4 in maize agronomic traits, we inserted the full-length cDNA of ZmDWF4 from the elite inbred Q319 into the expression vector pPZP211 driven by the maize ubiquitin promoter (Fig. 1a). Transgenic plants were obtained by an Agrobacterium-mediated transformation (Ishida et al. 2007) using the resistance gene NPTII as the selected marker for transgenic screening. Overall, we obtained twenty-two ZmDWF4 overexpression (ZmDWF4-OE) transgenic events, and three positive lines L100, L166, and L225 with high transgene expression levels were chosen for further analysis (Fig. 1b, c). These transgenic lines were further confirmed as independent transgenic events by southern blotting. The specific hybridization signal of both endogenous and transgenic ZmDWF4 gene was detected in the transgenic lines L100, L166 and L255 (Fig. 1f). These results indicated that T-DNA was stably inserted as a single copy into the genomes of these transgenic lines. Western blotting confirmed the elevated ZmDWF4 protein levels in the three lines (Fig. 1e). To detect BR levels, the contents of endogenous BL and CS in the leaves of the ZmDWF4-OE transgenic line L100 and wild type were measured using liquid chromatography-mass spectrography (LC-MS). Endogenous BL was not detected in the transgenic line L100 and wild type, consistent with the previous report by Wu et al. (2008). However, the endogenous CS level was dramatically increased by 92.6% in the transgenic line L100 compared with wild type (Fig. 1d).

Molecular identification of ZmDWF4 overexpression transgenic lines. a T-DNA structure of the vector pPZP211-Ubipro:ZmDWF4. Ubipro, the maize ubiquitin promoter; CaMV35Spro, the cauliflower mosaic virus 35S promoter; NPTII, the neomycin phosphotransferase gene. b PCR analysis for the genes NPTII or ZmDWF4 in transgenic lines. NC, no template control; PC, plasmid pPZP211-Ubipro:ZmDWF4 positive control; M, DL2000 DNA marker. c qPCR analysis for ZmDWF4 gene in transgenic lines and wild-type controls. d The content of the BR precursor canosterone (CS) in transgenic and wild-type plants. e Western blot analysis for ZmDWF4 protein in transgenic lines and wild-type controls. WT, non-transgenic Q319 control. f Southern blot analysis of the ZmDWF4 gene in transgenic lines and wild-type controls. WT, non-transgenic Q319 control. Means with the same letter are not significantly different at the 0.05 threshold using Tukey’s HSD test
ZmDWF4 expression promotes grain yield
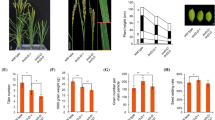
To assess whether the enhanced expression of ZmDWF4 could increase grain yield, field trials were conducted using T4 generations of ZmDWF4-OE lines and wild-type plants in 2015, and agronomic traits were determined with three replicates. As shown in Fig. 2 a and d, the ear length of the transgenic lines was significantly increased in line L100 (181.9 ± 9.5 cm), L166 (193.2 ± 14.2 cm), and L225 (184.6 ± 6.4 cm) by 14.4%, 21.4%, and 16.0%, respectively, compared with the ear length in the wild-type inbred Q319 (159.1 ± 10.8 cm); the ear diameter was significantly increased by 10.5%, 9.5%, and 13.4%, respectively (Fig. 2b, e), and the row number per ear was also increased by two rows in transgenic lines (Fig. 2c, Fig. S2b). Moreover, the kernel number per row was increased by 5 kernels (Fig. S2a) and there was a ~ 15% increase in the kernel number per ear in transgenic lines compared with wild-type ears (Fig. S2c). For analyzing changes in kernel size, we measured the length, width, and thickness of the kernels. The results showed that the kernel length of transgenic lines was significantly increased by up to 12.5% in ZmDWF4-OE lines (Fig. S2d) while the kernel width was increased more than 10% compared with the wild type (Fig. S2e). The kernel thickness of transgenic plants was not changed, and the kernel thickness of L100 was slightly reduced but not significantly (Fig. S2f). The 1000-grain weight of transgenic plants was also increased (Fig. 2f). Moreover, the grain weight per ear of ZmDWF4-OE transgenic lines was increased to about 28.3% to 33.5% more than that of wild-type controls (Fig. 2g).

Agronomic traits of ZmDWF4-overexpressing (ZmDWF4-OE) transgenic inbred lines in the field in 2015. a Ear phenotypes of ZmDWF4-OE lines and wild-type plants harvested at full maturity. b Transections of ears showing the kernel row number in transgenic and wild-type plants. c Seed phenotypes of transgenic and wild-type plants. d Ear length of ZmDWF4-OE maize compared with wild-type plants. e Ear diameter of ZmDWF4-OE transgenic lines and wild-type plants. f 1000-grain weight of ZmDWF4-OE maize and wild-type plants. g Grain weight per ear of ZmDWF4-OE maize compared with wild-type plants. Values are means of ten replicates ± SD. Means with the same letter are not significantly different at the 0.05 threshold using Tukey’s HSD test
Overexpressing ZmDWF4 enhances heterosis and grain yield of hybrids
To determine whether the overexpression of ZmDWF4 gene promotes grain yield in the transgenic hybrids, we used the ZmDWF4-OE transgenic inbred lines L100, L166, and L225, which were crossed to four commercially elite maize inbred lines in China, including two inbred lines of the Huanggai group (inbred 9801, 3841), one inbred line of Lancaster group (inbred V3B1), and one inbred line of the modified Reid group (inbred Z58) to generate F1 hybrids. For field trails, 67,500 F1 hybrid plants per hectare were grown in Tai’an in 2016. As shown in Fig. S1 and Table S1, these ZmDWF4 transgenic hybrids exhibited evidently improved both vegetative growth and yield traits compared with their corresponding non-transgenic hybrids control, such as increased ear length, ear diameter, kernel number, and kernel weight per ear. Among them, four hybrid combinations (ZmDWF4-OE×9801, ZmDWF4-OE×3841, ZmDWF4-OE×V3B1, and ZmDWF4-OE×Z58) showed more significant improvements in agronomic traits. As shown in Table 1, the ear length was increased between 6.4 (3.1%) and 15.7 cm (7.6%) (in ZmDWF4-OE×9801 hybrid), between 7.9 (3.9%) and 16.4 cm (8.3%) (in ZmDWF4-OE×3841 hybrid), between 8.2 (4.0%) and 13.3 cm (6.5%) (in ZmDWF4-OE×V3B1 hybrid), and between 12.5 (6.2%) and 39.8 cm (20.4%) (in ZmDWF4-OE×Z58 hybrid), respectively, in the transgenic hybrids compared with non-transgenic control hybrids. The ear diameter was also enhanced in the transgenic hybrids. The kernel row number was increased from 14 to 16 only in ZmDWF4-OE×3841 hybrid. The kernel number per row was also increased between 6.0 and 15.8% in the transgenic hybrids. Consequently, the seed number per ear was markedly improved between 2.4 and 24.2% in the transgenic hybrids compared with non-transgenic control hybrids. In addition to the increased kernel size and 1000-grain weight, the grain weight per ear was ultimately increased between 17.3 and 40.4%.
Overexpressing ZmDWF4 improves vegetative development and photosynthetic ability
Next, we examined agronomic traits in the transgenic plants. As expected, a number of agronomic traits were significantly improved, such as increased plant height, node number, and leaf area, delayed leaf senescence, and enhanced photosynthetic ability (Fig. 3 and S3). At the seedling stage, the transgenic lines grew faster than wild-type plants (Fig. 3a). At the six-leaf stage, the plant height and leaf area of transgenic plants were dramatically increased. As shown in Fig. 3d, the fourth leaf area increased by 26 cm2 (48.9%), the fifth leaf area increased by 37 cm2 (43.6%), and the sixth leaf area by 36 cm2 (22.4%) on average in transgenic plants compared with wild-type plants. At 10 DAP, the ZmDWF4-OE transgenic lines showed significant differences in many agronomic traits. Plant height increased by 26.0 to 53.0% in transgenic lines compared with wild-type plants (Fig. 3b, g) and the nodes number was increased from 16 in the wild type to 19 in the transgenic line L100 (Fig. 3c, e). In addition, ear leaf (EL) area increased by 235 cm2, 190 cm2, and 64 cm2 in the transgenic lines L100, L166, and L225, respectively. Similarly, the upper ear leaf (UEL) area and the lower ear leaf (LEL) area increased up to 40% compared with the corresponding areas of wild-type plants (Fig. 3h, i).

Phenotypic analysis of ZmDWF4-overexpressing (ZmDWF4-OE) transgenic plants. a Young seedlings of ZmDWF4-OE transgenic lines and wild-type Q319. b Phenotype of transgenic and wild-type plants at 10 days after pollination (DAP). c The stems of ZmDWF4-OE L100 and wild-type plants. Arrows point to the nodes. d Leaf area of the fourth, fifth, and sixth leaves of transgenic and wild-type plants at the six-leaf stage. e The number of nodes in mature transgenic and wild-type plants. f Chlorophyll index, relative to 0 DAP, of transgenic and wild-type plants. g Plant height of transgenic and wild-type plants. h, i The area of the UEL, the EL, and the LEL in transgenic and wild-type plants. Values are means of ten replicates ± SD. d, e, g, i Means with the same letter are not significantly different at the 0.05 threshold using Tukey’s HSD test. UEL, upper ear leaf; EL, the ear leaf; LEL, the lower ear leaf
Photosynthetic performance of the transgenic lines was also improved. The net photosynthesis rates were significantly increased in transgenic lines compared with that in the wild type at the 10 DAP (Fig. S3a, b). The chlorophyll index of the leaves of both transgenic lines and wild-type plants decreased over time after 10 DAP; however, the decrease was faster in wild type plants than in ZmDWF4-OE plants (Fig. 3f). This might have resulted from the delayed leaf senescence in transgenic lines. Thus, we evaluated the leaf senescence by examining the expression of the senescence marker genes ZmSEE1 (Zm00001d020636) at different time periods after pollination. After peaking at 20 DAP, transcriptional levels of ZmSEE1 decreased slower in the transgenic lines than in wild-type plants (Fig. S3c), indicating that the overexpressing ZmDWF4 delays the senescence of transgenic plants.
The analysis of seed wide-genome expression profiles of transgenic plants
Next, we examined gene expression in the kernels. We generated RNA-seq libraries of seeds at 10 and 15 DAP in the ZmDWF4-OE transgenic line L100 and wild-type plants, respectively. Forty genes randomly selected from the pool of DEGs were detected using qPCR to monitor the reliability of the RNA-seq data, and qPCR results were consistent with RNA-seq results (Fig. S4). At 10 DAP, there were 447 DEGs, and at 15 DAP, there were 645 DEGs; 117 DEGs were common between 10 DAP and 15 DAP (Fig. 4a). Volcano plots of the DEGs showed that activated genes outnumber the repressed genes (Fig. 4b, c). The significant enriched GO terms showed that the most significant enrichments were involved in innate immune response, nucleus importing, RNA synthesis and transporting, developmental process regulation, cell death, and defense response signaling pathway (Fig. 4d, e). Further, based on the Kyoto encyclopedia of Genes and Genomes (KEGG) pathway analysis, 53 DEGs in both 10 DAP data and 15 DAP data were mainly involved in 32 kinds of early seed development processes, which could be divided into five categories: cell growth and cell division, stress and immune response, hormone-related progress, nutrient reservoir activities, transport related processes (Fig. 4f). Most of DEGs had similar expression changes at both 10 DAP and 15 DAP. Notably, a great majority of DEGs involved in cell growth and cell division were upregulated. To assess the effect of ZmDWF4-OE on grain filling, we tabulated DEGs of transcription factors involved in starch synthesis. There were 10 DEGs and 12 DEGs at 10 and 15 DAP, respectively (Table S2). Meanwhile, we examined the expression levels of 2 genes in kernels, ZmCYP85A and ZmDET2, which were involved in BRs biosynthesis and signal pathways by qPCR at three stage (5, 10 and 15 DAP) of the transgenic line L100 and wild-type plants (Fig. S5). The transcriptional levels of ZmCYP85A and ZmDET2 were significantly increased to higher levels compared with wild-type plants. Overall, these results indicated that overexpressing ZmDWF4 gene affected both the BRs biosynthesis and signaling pathway. Taken together, these results indicated that overexpressing ZmDWF4 promotes the expression of the genes regulating cell division and storage reservoir, thus promoting endosperm development and grain filling, leading to the significantly increased seed yield of transgenic plants.

Distribution and classification of differentially expressed genes in the seed RNA-seq expression analysis. a Number of differentially expressed genes (DEGs) and their overlap at 10 and 15 DAP. b, c A volcano plot of expressed genes; red points, upregulated; green points, downregulated; black points, not differentially expressed. FC, fold change (L100/WT), FDR, false discovery rate. d, e GO enrichment analysis of differentially expressed genes (DEGs), the X-axis represents the negative logarithm (base 10) of the corrected P value. f Heatmaps of genes differentially expressed in both 10 DAP and 15 DAP seed RNA-seq data involved in five selected Kyoto Encyclopedia of Genes and Genomes pathways
Discussion
BRs signaling pathway plays a pivotal role in regulating plant growth and development (Best et al. 2016; Choe et al. 2001; Koh et al. 2007; Li et al. 2016; Makiko et al. 2003; Ming et al. 2007; Tomoaki et al. 2005; Wu et al. 2008). The Arabidopsis DWF4 gene encodes the sterol C-22 hydroxylase which catalyzes the key step in the BR biosynthesis pathway (Choe et al. 1998). In this study, we generated ZmDWF4 maize overexpression lines and found that elevated BR levels lead to a remarkably increased grain yield of both transgenic inbred corn plants and the hybrid combinations. Morphological and transcriptomic analysis revealed that BRs promoted development of agronomic trait, such as increased nodes number, plant height, leaf area, photosynthesis rate, length and row number of ears, as well as relative gene expression. Our results suggest a promising approach for high-yield breeding in maize by modification BR levels through transgenic biotechnology.
In terms of crop genetic improvement, the use of different regulatory elements might result in varying degrees of effectiveness for its purpose. Previously, the AtCYP90B1 gene, encoding a sterol C-22 hydroxylase, when it was specifically expressed in the embryo or endosperm of rice, and transgenic rice seed weight is not significantly increased (Wu et al. 2008). However, overexpression of AtCYP90B1 gene driven by CaMV 35S promoter in Arabidopsis and tomato results in 59 to 95% increases in yield, respectively (Choe et al. 2001; Li et al. 2016). When AtCYP90B1, ZmCYP724B3, or OsCYP90B2 was expressed under the control of the pAS promoter in rice, which specifically active in vegetative tissues, the transgenic rice plants showed more tillers and increased seed production about 40%. To achieve the maximum stimulation of yield, we chose the ubiquitin promoter rather than spatiotemporal-specific promoters to drive ZmDWF4 expression. Overexpression of the ZmDWF4 gene in both vegetative and reproductive organs improves vegetative growth and increases seed number, as well as 1000-grain weight. Meanwhile, we observed that all three ZmDWF4-OE transgenic lines showed early flowering phenotype (Fig. S6). The earing time and heading time of ZmDWF4-OE transgenic lines were about five to 7 days earlier than that of wild-type plants. This might be resulted from improved vegetative growth in ZmDWF4-OE plants. Thus, we propose that the overexpression of ZmDWF4 in both vegetative and reproductive organs might promote seed-filling in transgenic plants. These results imply that simultaneous expansion of plant vegetative and reproductive tissues is an effective strategy for high-yield crop breeding by enhancing carbon fixation in the leaf and nutrition accumulation in the seed.
In agricultural practice of maize plantation, hybrid seeds are planted rather than inbred lines due to heterosis. Considering that genetic diversity of different maize germplasm has an impact on heterosis of the same parental inbred maize, we chose four elite inbred lines for mating to evaluate the transgene efficacy of ZmDWF4 overexpression on grain yield. In this study, we generated 16 hybrids using these four cultivars mated with non-transgenic Q319 and T4 plants of ZmDWF4-OE transgenic lines L100, L165, and L225. During field trials, all the transgenic hybrids showed increased grain yield compared with corresponding wild-type controls; however, the yield increasing was variable among hybrid combinations (Table 1 and Fig. S1). The ear length was increased by 3.1 to 20.4%, and the kernel number per row was increased by 6.0 to 15.8% leading to an average final grain yield increase of more than 17.3%. Although the 1000-grain weight was not significantly elevated in the transgenic inbred Q319 plants, the grain size was significantly larger in transgenic hybrids. As reported in early maize ear inflorescence development, the expression patterns of many transcription factors in BR signaling pathways are changed, such as the ZmBZR1 gene, which is upregulated in F1 hybrid compared with its Mo17 parent (Ding et al. 2014). We suggested that the increased BR levels enhance heterosis not only in terms of ear architectural traits but also in terms of kernel size. For the limitation of field trial (just 1 year and one location), the extent of yield increase might be slightly inaccurate and multi-year/location field trials are needed to further confirm the increase extent of grain yield. Our results show that enhancing endogenous BR levels can effectively increase grain yield in hybrids maize plants containing the overexpressing ZmDWF4. Such transgenic lines might be valuable for further high-yield maize breeding.
References
Best NB, Hartwig T, Budka J, Fujioka S, Johal G, Schulz B, Dilkes BP (2016) nana plant2 encodes a maize ortholog of the Arabidopsis brassinosteroid biosynthesis gene DWARF1, identifying developmental interactions between brassinosteroids and gibberellins. Plant Physiol 171:2633–2647. https://doi.org/10.1104/pp.16.00399
Choe S, Dilkes BP, Fujioka S, Takatsuto S, Sakurai A, Feldmann KA (1998) The DWF4 gene of Arabidopsis encodes a cytochrome P450 that mediates multiple 22α-hydroxylation steps in brassinosteroid biosynthesis. Plant Cell 10:231–243. https://doi.org/10.1105/tpc.10.2.231
Choe S, Fujioka S, Noguchi T, Takatsuto S, Yoshida S, Feldmann KA (2001) Overexpression of DWARF4 in the brassinosteroid biosynthetic pathway results in increased vegetative growth and seed yield in Arabidopsis. Plant J 26:573–582. https://doi.org/10.1046/j.1365-313x.2001.01055.x
Choudhary SP, Yu J-Q, Yamaguchi-Shinozaki K, Shinozaki K, Tran L-SP (2012) Benefits of brassinosteroid crosstalk. Trends Plant Sci 17:594–605. https://doi.org/10.1016/j.tplants.2012.05.012
Ding H, Qin C, Luo X, Li L, Chen Z, Liu H, Gao J, Lin H, Shen Y, Zhao M, Lübberstedt T, Zhang Z, Pan G (2014) Heterosis in early maize ear inflorescence development: a genome-wide transcription qnalysis for two maize inbred lines and their hybrid. Int J Mol Sci 15:13892–13915. https://doi.org/10.3390/ijms150813892
Feng Z, Wu C, Wang C, Roh J, Zhang L, Chen J, Zhang S, Zhang H, Yang C, Hu J, You X, Liu X, Yang X, Guo X, Zhang X, Wu F, Terzaghi W, Kim S-K, Jiang L, Wan J (2016) SLG controls grain size and leaf angle by modulating brassinosteroid homeostasis in rice. J Exp Bot 67:4241–4253. https://doi.org/10.1093/jxb/erw204
Fridman Y, Savaldi-Goldstein S (2013) Brassinosteroids in growth control: how, when and where. Plant Sci 209:24–31. https://doi.org/10.1016/j.plantsci.2013.04.002
Fujioka S, Noguchi T, Yokota T, Takatsuto S, Yoshida S (1998) Brassinosteroids in Arabidopsis thaliana. Phytochemistry 48:595–599. https://doi.org/10.1016/S0031-9422(98)00065-X
Fujita S, Ohnishi T, Watanabe B, Yokota T, Takatsuto S, Fujioka S, Yoshida S, Sakata K, Mizutani M (2006) Arabidopsis CYP90B1 catalyses the early C-22 hydroxylation of C27, C28 and C29 sterols. Plant J 45:765–774. https://doi.org/10.1111/j.1365-313X.2005.02639.x
Guo H, Li L, Aluru M, Aluru S, Yin Y (2013) Mechanisms and networks for brassinosteroid regulated gene expression. Curr Opin Plant Biol 16:545–553. https://doi.org/10.1016/j.pbi.2013.08.002
Hannah LC, Futch B, Bing J, Shaw JR, Boehlein S, Stewart JD, Beiriger R, Georgelis N, Greene T (2012) A shrunken-2 transgene increases maize yield by acting in maternal tissues to increase the frequency of seed development. Plant Cell 24:2352–2363. https://doi.org/10.1105/tpc.112.100602
Hartwig T, Chuck GS, Fujioka S, Klempien A, Weizbauer R, Potluri DPV, Choe S, Johal GS, Schulz B (2011) Brassinosteroid control of sex determination in maize. Proc Natl Acad Sci U S A 108:19814–19819. https://doi.org/10.1073/pnas.1108359108
Ishida Y, Hiei Y, Komari T (2007) Agrobacterium-mediated transformation of maize. Nat Protoc 2:1614–1621. https://doi.org/10.1038/nprot.2007.241
Kir G, Ye H, Nelissen H, Neelakandan AK, Kusnandar AS, Luo A, Inzé D, Sylvester AW, Yin Y, Becraft PW (2015) RNA interference knockdown of BRASSINOSTEROID INSENSITIVE1 in maize reveals novel functions for brassinosteroid signaling in controlling plant architecture. Plant Physiol 169:826–839. https://doi.org/10.1104/pp.15.00367
Koh S, Lee S-C, Kim M-K, Koh JH, Lee S, An G, Choe S, Kim S-R (2007) T-DNA tagged knockout mutation of rice OsGSK1, an orthologue of Arabidopsis BIN2, with enhanced tolerance to various abiotic stresses. Plant Mol Biol 65:453–466. https://doi.org/10.1007/s11103-007-9213-4
Li J, Nagpal P, Vitart V, McMorris TC, Chory J (1996) A role for brassinosteroids in light-dependent development of Arabidopsis. Science 272:398–401. https://doi.org/10.1126/science.272.5260.398
Li B, Liu H, Zhang Y, Kang T, Zhang L, Tong J, Xiao L, Zhang H (2013) Constitutive expression of cell wall invertase genes increases grain yield and starch content in maize. Plant Biotechnol J 11:1080–1091. https://doi.org/10.1111/pbi.12102
Li W, Zhao X, Zhang X (2015) Genome-wide analysis and expression patterns of the YUCCA genes in maize. J Genet Genomics 42:707–710. https://doi.org/10.1016/j.jgg.2015.06.010
Li X-J, Guo X, Zhou Y-H, Shi K, Zhou J, Yu J-Q, Xia X-J (2016) Overexpression of a brassinosteroid biosynthetic gene Dwarf enhances photosynthetic capacity through activation of Calvin cycle enzymes in tomato. BMC Plant Biol 16:33. https://doi.org/10.1186/s12870-016-0715-6
Liu T, Zhang J, Wang M, Wang Z, Li G, Qu L, Wang G (2007) Expression and functional analysis of ZmDWF4, an ortholog of Arabidopsis DWF4 from maize (Zea mays L.). Plant Cell Rep 26:2091–2099. https://doi.org/10.1007/s00299-007-0418-4
Liu X, Feng ZM, Zhou CL, Ren YK, Mou CL, Wu T, Yang CY, Liu SJ, Jiang L, Wan JM (2016) Brassinosteroid (BR) biosynthetic gene lhdd10 controls late heading and plant height in rice (Oryza sativa L.). Plant Cell Rep 35:357–368. https://doi.org/10.1007/s00299-015-1889-3
Makiko C et al (2003) A semidwarf phenotype of barley uzu results from a nucleotide substitution in the dene encoding a putative brassinosteroid receptor. Plant Physiol 133:1209–1219. https://doi.org/10.1104/pp.103.026195
Ming L, Yuehua X, Xianbi L, Xiaofeng L, Wei D, Demou L, Lei H, Mingyu H, Yi L, Yan P (2007) GhDET2, a steroid 5α-reductase, plays an important role in cotton fiber cell initiation and elongation. Plant J 51:419–430. https://doi.org/10.1111/j.1365-313X.2007.03144.x
Ren RC, Wang LL, Zhang L, Zhao YJ, Wu JW, Wei YM, Zhang XS, Zhao XY (2020) DEK43 is a P-type pentatricopeptide repeat (PPR) protein responsible for the Cis-splicing of nad4 in maize mitochondria. J Integr Plant Biol 62:299–313. https://doi.org/10.1111/jipb.12843
Shimada S, Komatsu T, Yamagami A, Nakazawa M, Matsui M, Kawaide H, Natsume M, Osada H, Asami T, Nakano T (2015) Formation and dissociation of the BSS1 protein complex regulates plant development via brassinosteroid signaling. Plant Cell 27:375–390. https://doi.org/10.1105/tpc.114.131508
Si J, Sun Y, Wang L, Qin Y, Wang C, Wang X (2016) Functional analyses of Populus euphratica brassinosteroid biosynthesis enzyme genes DWF4 (PeDWF4) and CPD (PeCPD) in the regulation of growth and development of Arabidopsis thaliana. J Biosci 41:727–742. https://doi.org/10.1007/s12038-016-9635-8
Singh AP, Savaldi-Goldstein S (2015) Growth control: brassinosteroid activity gets context. J Exp Bot 66:1123–1132. https://doi.org/10.1093/jxb/erv026
Singh A, Breja P, Khurana JP, Khurana P (2016) Wheat Brassinosteroid-Insensitive1 (TaBRI1) interacts with members of TaSERK gene family and cause early flowering and seed yield enhancement in Arabidopsis. PLoS One 11:e0153273. https://doi.org/10.1371/journal.pone.0153273
Sun X, Cahill J, Van Hautegem T, Feys K, Whipple C, Novák O, Delbare S, Versteele C, Demuynck K, De Block J, Storme V, Claeys H, Van Lijsebettens M, Coussens G, Ljung K, De Vliegher A, Muszynski M, Inzé D, Nelissen H (2017) Altered expression of maize PLASTOCHRON1 enhances biomass and seed yield by extending cell division duration. Nat Commun 8:14752. https://doi.org/10.1038/ncomms14752
Szekeres M, Németh K, Koncz-Kálmán Z, Mathur J, Kauschmann A, Altmann T, Rédei GP, Nagy F, Schell J, Koncz C (1996) Brassinosteroids rescue the deficiency of CYP90, a cytochrome P450, controlling cell elongation and de-etiolation in Arabidopsis. Cell 85:171–182. https://doi.org/10.1016/S0092-8674(00)81094-6
Tian Y, Fan M, Qin Z, Lv H, Wang M, Zhang Z, Zhou W, Zhao N, Li X, Han C, Ding Z, Wang W, Wang Z-Y, Bai M-Y (2018) Hydrogen peroxide positively regulates brassinosteroid signaling through oxidation of the BRASSINAZOLE-RESISTANT1 transcription factor. Nat Commun 9:1063–1063. https://doi.org/10.1038/s41467-018-03463-x
Tomoaki S, Yoichi M, Toshiyuki O, Hidehiko S, Shozo F, Miyako U-T, Masaharu M, Kanzo S, Suguru T, Shigeo Y, Hiroshi T, Hidemi K, Makoto M (2005) Erect leaves caused by brassinosteroid deficiency increase biomass production and grain yield in rice. Nat Biotechnol 24:105–109. https://doi.org/10.1038/nbt1173
Tong H, Xiao Y, Liu D, Gao S, Liu L, Yin Y, Jin Y, Qian Q, Chu C (2012) DWARF AND LOW-TILLERING acts as a direct downstream target of a GSK3/SHAGGY-like kinase to mediate brassinosteroid responses in rice. Plant Cell 24:2562–2577. https://doi.org/10.1105/tpc.112.097394
Tong H, Liu L, Jin Y, Du L, Yin Y, Qian Q, Zhu L, Chu C (2014) Brassinosteroid regulates cell elongation by modulating gibberellin metabolism in rice. Plant Cell 26:4376–4393. https://doi.org/10.1105/tpc.114.132092
Vogler F, Schmalzl C, Englhart M, Bircheneder M, Sprunck S (2014) Brassinosteroids promote Arabidopsis pollen germination and growth. Plant Reprod 27:153–167. https://doi.org/10.1007/s00497-014-0247-x
Wang X, Zhang J, Yuan M, Ehrhardt DW, Wang Z, Mao T (2012) Arabidopsis MICROTUBULE DESTABILIZING PROTEIN40 is involved in brassinosteroid regulation of hypocotyl elongation. Plant Cell 24:4012–4025. https://doi.org/10.1105/tpc.112.103838
Wei Z, Li J (2016) Brassinosteroids regulate root growth, development, and symbiosis. Mol Plant 9:86–100. https://doi.org/10.1016/j.molp.2015.12.003
Wu C-y, Trieu A, Radhakrishnan P, Kwok SF, Harris S, Zhang K, Wang J, Wan J, Zhai H, Takatsuto S, Matsumoto S, Fujioka S, Feldmann KA, Pennell RI (2008) Brassinosteroids regulate grain filling in rice. Plant Cell 20:2130–2145. https://doi.org/10.1105/tpc.107.055087
Wu J, Lawit SJ, Weers B, Sun J, Mongar N, Van Hemert J, Melo R, Meng X, Rupe M, Clapp J, Haug Collet K, Trecker L, Roesler K, Peddicord L, Thomas J, Hunt J, Zhou W, Hou Z, Wimmer M, Jantes J, Mo H, Liu L, Wang Y, Walker C, Danilevskaya O, Lafitte RH, Schussler JR, Shen B, Habben JE (2019) Overexpression of zmm28 increases maize grain yield in the field. Proc Natl Acad Sci U S A 116:23850–23858. https://doi.org/10.1073/pnas.1902593116
Xia XJ, Zhou YH, Shi K, Zhou J, Foyer CH, Yu JQ (2015) Interplay between reactive oxygen species and hormones in the control of plant development and stress tolerance. J Exp Bot 66:2839–2856
Xie G, Li Z, Ran Q, Wang H, Zhang J (2018) Over-expression of mutated ZmDA1 or ZmDAR1 gene improves maize kernel yield by enhancing starch synthesis. Plant Biotechnol J 16:234–244. https://doi.org/10.1111/pbi.12763
Xin P, Li B, Yan J, Chu J (2018) Pursuing extreme sensitivity for determination of endogenous brassinosteroids through direct fishing from plant matrices and eliminating most interferences with boronate affinity magnetic nanoparticles. Anal Bioanal Chem 410:1363–1374. https://doi.org/10.1007/s00216-017-0777-9
Zhang C, Xu Y, Guo S, Zhu J, Huan Q, Liu H, Wang L, Luo G, Wang X, Chong K (2012) Dynamics of brassinosteroid response modulated by negative regulator LIC in rice. PLoS Genet 8:e1002686. https://doi.org/10.1371/journal.pgen.1002686
Zhang C, Bai M-Y, Chong K (2014a) Brassinosteroid-mediated regulation of agronomic traits in rice. Plant Cell Rep 33:683–696. https://doi.org/10.1007/s00299-014-1578-7
Zhang WY, Xu YC, Li WL, Yang L, Yue X, Zhang XS, Zhao XY (2014b) Transcriptional analyses of natural leaf senescence in maize. PLoS One 9:e115617. https://doi.org/10.1371/journal.pone.0115617
Zhao XY, Hong P, Wu JY, Chen XB, Ye XG, Pan YY, Wang J, Zhang XS (2016) The tae-miR408-mediated control of TaTOC1 genes transcription is required for the regulation of heading time in wheat. Plant Physiol 170:1578–1594. https://doi.org/10.1104/pp.15.01216
Funding
This research was supported by the Major Project of China on New varieties of GMO Cultivation (2016ZX08003-003), the National Natural Science Foundation of China (91735301 and 91535109), Taishan Scholars Project (ts201712024), Funds of Shandong “Double Tops” Program (SYL2017YSTD03), and the project (dxkt201707) from State Key Laboratory of Crop Biology.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 3904 kb)
Rights and permissions
About this article

Cite this article
Liu, N., Zhao, Y.J., Wu, J.W. et al. Overexpression of ZmDWF4 improves major agronomic traits and enhances yield in maize. Mol Breeding 40, 71 (2020). https://doi.org/10.1007/s11032-020-01152-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-020-01152-6