
Abstract
The aim of this study was to perform genome-wide selection using a set of Dart-seq markers associated to the additive-dominant genomic best linear unbiased prediction (GBLUP) model to predict maize grain yield in different crop seasons and locations. Genotyping was performed with Dart-seq markers from 447 lines coming from a germplasm bank of a private maize breeding company. Crossing these lines provided 838 single-cross hybrids evaluated in six locations in the winter crop season of 2013 and 797 single-cross hybrids evaluated in four locations in the summer crop season of 2013/2014. Four k-fold levels were applied on the full panel of 23,153 Dart genotyping-by-sequencing markers and samples of 50% of the available markers. The different crop seasons were used as training and validation populations to estimate the predictive accuracy. The magnitude of the correlations between predicted and observed hybrids ranged from 0.82 to 0.89 in the winter crop season and from 0.56 to 0.76 in the summer crop season. The correlations between combinations tested in different crop seasons and locations were encouraging (0.53). Predictive ability was highly influenced by the genetic background and also by the interaction between crop seasons. The coincidences between the genomic values of the summer crop and winter crop, in terms of discard, were 89 and 90%. This result shows the possibility of using genomic prediction in breeding programs for initial discard of low-yielding genotypes. The GBLUP method was able to generate high correlations between predicted and observed hybrids, even at high levels of missing in k-fold and in different locations and crop seasons.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Maize breeding programs aim to develop high-yielding hybrids that are better than those already in the market. However, achieving this objective is not simple. Maize breeding involves systems of crosses that aim to identify the best combinations of parents where the hybrids are the final product (Hallauer et al. 2010). To test all possible combinations of hybrids, it would be necessary to perform thousands of crosses, which becomes unfeasible in breeding programs. For this reason, only part of the crosses is carried out, thus reducing the chances of finding the most promising combinations (Schrag et al. 2010).
With advancement in the use of molecular markers, the selection for important traits such as grain yield, which was originally performed on by phenotypic information, can now be performed using the molecular marker’s information. A justification for the use of molecular markers is the expectation that information on genomic constitution of the individuals may bring greater genetic gain than when only phenotypic data are used (Meuwissen et al. 2001).
Combining the information from molecular markers and phenotypic data, a model for prediction of maize hybrids was proposed by Bernardo (1994) using best linear unbiased prediction (BLUP), which takes the information from molecular markers into consideration for construction of the pedigree of the parental lines. Although the correlation between the predicted genotypic performance and its phenotypic value is moderate in most cases, there were advantages to this approach (Bernardo 1994). This approach proved to be effective in various other studies (Bernardo 1995, 1996a, b; Massman et al. 2013; Cantelmo et al. 2016).
Nevertheless, with the increase in the availability of markers available and reduction in costs of genotyping, the techniques of genome-wide selection (GWS) became a reality for breeding programs (Heslot et al. 2014; Zhao et al. 2015). Genomic prediction has proven to be an alternative tool that uses all the information from molecular markers for selection of the best combinations, allowing the breeder to obtain gains per unit time. In contrast with using only the molecular markers that exhibit significance or linkage disequilibrium with quantitative trait loci (QTL), genome-wide prediction uses all the markers simultaneously in the linear model (Windhausen et al. 2012).
Meuwissen et al. (2001) were the first to incorporate a large number of markers in the genomic model for prediction of genetic values. The GWS proposed by these authors simultaneously considers all the markers distributed over the genome in a prediction model, incorporating the information from large- and small-effect QTL in genetic variance. In this scenario, each marker is considered a possible QTL which, together, is combined to predict the genetic value of the genotype (Guo et al. 2013a).
Among the models used for GWS, the genomic best linear unbiased prediction (GBLUP) model, proposed by VanRaden (2008), has been widely used in plant breeding (Crossa et al. 2013). The GBLUP uses genomic covariance to estimate the genetic merit of an individual. For that purpose, a genomic relationship matrix estimated from information from molecular markers is used to recover information in related individuals (Crossa et al. 2013; Hayes et al., 2009a, b; VanRaden 2008). Results indicate advantages of the GBLUP model in relation to marker models due to its relative simplicity and shorter computational time, which, in addition to the already well-known properties of the mixed models of selection, makes this a very attractive approach from the genetic and statistical perspective (Heslot et al. 2014; VanRaden et al. 2009).
However, most studies described in the literature have used the GBLUP that includes only the additive effects (Calus 2010), and only a reduced number of studies have taken the dominance effects into consideration (Vitezica et al. 2013; Santos et al. 2016). Thus, more recently, the GBLUP including dominance effects has been suggested to improve the prediction of missing hybrids, especially when there is a pronounced dominance effect in the trait (Nishio and Satoh 2014).
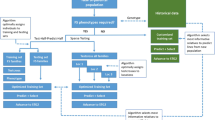
In genomic selection, the predictive ability of these models has been evaluated by cross-validation methods, which has somewhat overshadowed the evaluation of the breeder regarding the real efficacy of the method. In this respect, it becomes necessary to evaluate the predictive ability of this technique in a real situation of prediction and validation in different crop seasons and environments. Therefore, the aim of this study was to apply genome-wide prediction for maize single-cross hybrids using a set of Dart genotyping-by-sequencing (GBS) markers associated with the additive-dominant GBLUP model. We also sought to evaluate the predictive ability of this model through cross-validation and real validation using one crop season as a training population and another crop season with different locations as validation.
Materials and methods
Obtaining single-cross hybrids and phenotypic evaluation
The genetic material used consisted of 447 lines from different backgrounds from various regions of the world, which were used in crosses to generate single-cross hybrids.
Crossing these lines provided 838 single-cross hybrids evaluated in the winter crop season of 2013 and 797 single-cross hybrids evaluated in the summer crop season of 2013/2014. In the winter crop season, the maize single-cross hybrids were evaluated in six locations: Primavera do Leste, MT; Sorriso, MT; Rio Verde, GO; Lucas do Rio Verde, MT; Campo Novo do Parecis, MT; and Sapezal, MT, located in the central south region of Brazil. In the summer crop season, the hybrids were evaluated in four locations: Araguari, Nova Ponte, Presidente Olegário, and Uberlândia, all located in the state of Minas Gerais, Brazil.
The experiments were carried out using an incomplete block experimental design with two replications per location; plots were composed of four 5-m-length rows and 0.7 m between rows.
Grain yield of the hybrids was evaluated and corrected to 13% moisture content and converted to tons per hectare. The area was tilled and top-dressed fertilizer was applied according to recommendations for each experimental area. Crop treatments were conducted for control of fall armyworm (Spodoptera frugiperda) and corn earworm (Helicoverpa zea), as well as for weed control.
Grain yield data of all the hybrids in all the experiments were analyzed using mixed models with the following model:
where X corresponds to the fixed effects matrix: replication within experiment within location, experiment within location, and a location; β is the incidence vector of fixed effects; Z is the random effects matrix: block within replication within experiment and within location; b is the incidence vector of the effects described in Z; W is the matrix of genotypes; g is the incidence vector of genotypes; T is the matrix of the genotype × environment interaction; i is the incidence vector of the genotype × environment interaction; and ε is the residual vector.
The estimates of the fixed effects, the phenotypic variance components, and the predictions of the random effects were obtained through restricted maximum likelihood (REML) using the expectation-maximization (EM) algorithm. Broad-sense heritability of the analyses between the environments was calculated using the equation: \( {h}^2={\sigma}_{\mathrm{g}}^2/\left({\sigma}_{\mathrm{g}}^2+{\sigma}_{\mathrm{g}\mathrm{xe}}^2/ e+{\sigma}_r^2/ re\right) \), in which \( {\sigma}_{\mathrm{g}}^2 \), \( {\sigma}_{\mathrm{gxe}}^2 \), and \( {\sigma}_r^2 \) correspond to genotypic variance, variance of the genotype × environment interaction, and residual variance, respectively, and r corresponds to the number of replications and e to the number of environments (Hallauer et al. 2010). This analysis was performed using the procedure Proc Mixed on the SAS platform.
Genotyping and construction of the marker matrix
DNA extraction was carried out following the specific protocol of the Diversity Arrays Technology Company (Vitezica et al. 2013) to which appropriately diluted DNA samples were sent for analysis. The 470 lines were genotyped using the Dart GBS markers; however, due to non-amplification of some samples sent, genotype data were obtained for 447 lines.
The information matrix of markers in the hybrids was constructed using code 1 for the presence of the allele t in marker m in parental line i, 0 for the absence of the allele, and “−” when reliable reading by the software was not possible. This codification facilitates the construction of additive matrices (2, 1, and 0) and dominant matrices of the hybrids contrary to the usual codification of 2 and 0 that codifies homozygote and diploid lines.
Missing markers were imputed by the function A.mat, mean method, of the rrBLUP package (Endelman 2011) of the R software.
Prediction using the GBLUP model
The genetic value prediction process of the single-cross hybrids evaluated in the first and second crop trials was carried out by the GBLUP model with inclusion of additive and dominance effects, which is defined by
where \( \tilde{\mathbf{y}} \) is the vector of the observations of the phenotypic means n × 1, corrected for fixed effects and random effects of blocks described in Eq. 1, and n corresponds to the total number of observations, given by \( n=\sum_{i=1}^t{n}_i \), in which n i is the number of single-cross hybrids evaluated in location i and t is the number of locations; X is the incidence matrix of the fixed effects of locations confounded with the general mean n × t; \( \widehat{\boldsymbol{\upbeta}} \) is the fixed effects vector of the general mean confounded with locations t × 1; Z is the incidence matrix of the single-cross hybrids evaluated in each location n × p; α is the additive effects vector p × 1; δ is the dominance effect vector p × 1; and e is the residual vector n × 1.
The matrix of molecular markers of the single-cross hybrids was obtained by the sum of the matrices of markers of the two previously genotyped parental lines.
The matrices of additive and dominance markers were corrected using the Cockerham (1954) metric as decrypted by Vitezica et al. (2013)
where p k is the frequency of the favorable allele in locus k, W α is the incidence matrix of the additive effects of the markers, and W δ is the incidence matrix of the dominance effects of the markers. This parametric approach allows the orthogonality between additive and non-additive effects. More details about the genetic justification can be obtained in Zeng et al. (2005) and Vitezica et al. (2013).
The additive and dominance relationship matrices were obtained according to Vitezica et al. (2013):
where A is the additive relationship matrix described in VanRaden (2008) and D is the dominance relationship matrix.
The distributions of the random effects are considered as α ∼ MVN(0, G α ), δ ∼ MVN(0, G δ ), and e ∼ MVN(0, R), in which \( {\mathbf{G}}_{\alpha}=\mathbf{A}{\widehat{\sigma}}_{\alpha}^2 \), \( {\mathbf{G}}_{\delta}=\mathbf{D}{\widehat{\sigma}}_{\delta}^2 \), and \( \mathbf{R}=\mathbf{I}{\widehat{\sigma}}_e^2 \). The observations are assumed as \( \tilde{\mathbf{y}}\sim MVN\left(0,\mathbf{V}\right) \), where V = ZG α Z ′ + ZG δ Z ′ + R.
Estimation of the variance components was obtained by REML based on Fisher’s scoring algorithm.
The solution for the fixed, additive, and dominance effects was obtained by
The estimates of the standard deviations of the variance components were obtained by \( {\mathbf{s}}_{\left({\sigma}^2\right)}=\sqrt{\operatorname{diag}\Big({\mathbf{I}}_o}\Big) \) in which I o is the expected information matrix obtained by the inverse of the negative of the Hessian matrix.
All processes of analysis were carried out on the platform R (Zhao et al. 2015). The R codes used for analysis (S1-Text) and molecular data (S2-Dart) are available in the supplemental material.
For the predictions among the genomic values (GVs) obtained in the winter and summer crop seasons, 262 lines corresponding to 395 hybrids common in both crop seasons were used. In this sense, 395 hybrids evaluated in both seasons were utilized as control in order to obtain a threshold for the predictive accuracy.
Cross-validation
The cross-validation method used was k-fold, in which k was calculated for different levels. The set of observations was randomly subdivided into data groups per location according to the k-fold percentage adopted. In the process of analysis, a group was eliminated sequentially and the remaining individuals were used to compose the training population. In prediction of the genetic values of the genotypes that were part of the validation group, two modifications were made in the structures of the X and Z matrices: (i) elimination of the lines of the X matrix related to the genotypes that were part of the validation group and (ii) elimination of the lines of the Z matrix related to the genotypes of the same group, such that Z ′ Z has the same dimension of complete G α and G δ , and the predicted hybrids were obtained by direct solution of α and δ.
The k-fold levels used were correspondent to 10, 20, 30, and 50% of missing hybrids, i.e., 10-fold, 20-fold, 30-fold, and 50-fold, respectively. Efficiency was estimated by the Pearson correlation between the predicted genetic values of the validation group and the estimated genetic values in analysis considering the groups. This correlation was used as a parameter of cross-validation. Each cross-validation process was repeated 100 times.
Prediction of all the hybrid combinations and validation in different crop seasons and locations
Considering all the 447 line combinations, it is possible to obtain 99,681 maize hybrids. Thus, the complete A and D matrices were constructed considering all the possible crosses between these lines in a complete diallel. Based on the results of the 2013 crop season, all the hybrid combinations not evaluated were predicted and, among these, 402 were evaluated in the summer crop season (2014). Thus, the 2013 crop season served as a training and prediction population of the 402 missing hybrids that were later evaluated in the 2013/2014 crop season. The accuracy of prediction was measured using the Pearson correlation between the predicted and observed values. Also, a proportion of selection, corresponding to 20% of the hybrids predicted in the 2013 crop season, was used and the percentage of coincidence for these same hybrids in the 2014 crop season was estimated. This index was used to verify the selective ability of the method. To verify the discard ability, the same index was applied, considering the 20% lowest-yielding hybrids.
Clustering heterotic groups
The additive relationship matrix (A) was created according to the proposal of Vitezica et al. (2013) to comprehend the genetic relationships between the parental lines
where p is the frequency of the favorable allele and W A is the centered matrix of the markers.
After that, hierarchical clustering was analyzed through the hclust function of the hclust package in the R software (Xua et al. 2014) configured by the Wald method, using a matrix of the Euclidean distance of the elements of the A matrix as an object.
Results
Genetic parameters and line clusters
The mean yield of the hybrids was 5.33 t ha−1 in the winter crop season and 8.21 t ha−1 in the summer crop season. Table 1 shows the genetic variance (\( {\sigma}_{\mathrm{g}}^2 \)), the genotype-by-environment variance (\( {\sigma}_{\mathrm{gxe}}^2 \)), and the residual variance (\( {\sigma}_r^2 \)), in addition to broad-sense heritability.
All the variance components were statistically different from zero by the asymptotic Z test. The significance of genotypic variance indicates the variability that exists between them, thus allowing selection of the most promising. The ratio between the variance of the genotype × environment interaction and genetic variance ranged from 0.99 for the winter crop season to 0.13 for the summer crop season. The ratio between the residual and genetic variances was stable between the two crop seasons, i.e., 3.94 for the winter crop season and 3.92 for the summer crop season.
Broad-sense heritability was similar between the two crop seasons, even though the summer crop season had fewer environments. This situation was compensated by its greater genetic variance and lower interaction variance, thus leading to a heritability of 0.66.
By genotypic analysis of the lines, 23,153 Dart GBS markers were obtained, distributed over nine linkage groups, mitochondria, plastids, and some with unknown positions. The number of markers ranged from 3612 in chromosome 1 to 1386 in chromosome 7 (Table 2). Markers were not mapped in the 10th chromosome by the Diversity Arrays Technology Company. The lines clustering based on the additive matrix showed that the majority of genotypes agree with prior background information, but some lines presented a different pattern than expected. For example, some flint lines were clustered as Stiff Stalk Synthetic.
Genomic analysis and cross-validation for summer and winter seasons
The results of GBLUP analysis show the dominance variance (DV) observed in the summer crop season was greater than the additive variance (AV). This superiority shows the predominance of the non-additive effects for the yield trait in this crop season. A greater genotype-by-environment interaction can also be observed when compared to the winter crop season.
The AV and DV showed similar values for the winter crop season. Comparing the variance components obtained by the REML mixed models method based on raw data and the model with markers with corrected phenotypic values, a good recovery of the genetic variance (Table 1) by means of the sums of the additive and dominance variances estimated from the GBLUP model was observed (Table 3).
Correlations in the summer crop season were lower than those observed in the winter crop season, ranging from 0.73 to 0.56. The lower correlation and higher variation can be explained due to the smaller dataset and greater genotype × environment interaction. A greater influence of the degree of missing data in this season can also be observed, such that for the greatest fold level (50%), there was a strong decline in predictive ability, which did not occur in the winter crop season (Tables 3 and 4).
A systematic sampling of 50% of the markers (11,576) was also used to investigate the decrease in the number of markers in prediction of hybrids. This sampling was carried out, keeping a homogeneous distribution of the markers over the genome. In genome-wide prediction, using cross-validation, the correlation between predicted and observed GVs in the winter crop season ranged from 0.89 to 0.82, with discrete reduction in correlation with an increase in the fold level. It should be noted that even with an unbalance of 50%, accuracies above 0.80 can be observed (Table 3).
The decrease in density of markers by half in the dataset of the winter crop season practically did not influence the predictive ability of the model. The correlation remained stable when the full or half panel of markers was used, even at the highest fold levels.
In relation to reduction in the number of molecular markers, it was possible to observe a low influence of missing data since the correlations were constant for the two scenarios (full and half panels).
Prediction validation across the winter and summer seasons
Considering both two crop seasons, 395 hybrids were taken as common. These genotypes were ranked according to their GV observed in each season, and an intensity of selection and discard of 20% was used. It was found that the response based on the winter season is not very effective to predict the best genotypes in the summer season (coincidence of 43%), but the screening results based on the discard of the worst lines were encouraging. In other words, discarding the 20% lowest-yielding hybrids, a discarding error of only 11% was observed; that is, only nine superior genotypes would be mistakenly discarded based on the GVs observed in the winter crop season, aiming the selection in the summer crop season.
Based on the phenotypic data of the winter crop season, the prediction of 98,790 hybrid combinations was possible (two parental lines were discarded). Of the predicted hybrids not evaluated in the winter crop season (training population), 402 were planted in the summer crop season (validation population). The correlation between the GVs predicted in the winter crop season and the mean values observed in the summer crop season was 0.53 (Fig. 1a). The discrepancy of the climatic conditions in the two crop seasons is noteworthy, as well as the different sets of environments used, located in different states and regions. This low prediction ability can be justified by the correlation between the performance of the 395 different hybrids evaluated in both crop seasons that was 0.60 (Fig. 1b).

Linear regression of the 402 predicted genomic values using the winter crop season as training population over their observed values in the summer crop season (a). Linear regression of the 395 different hybrids that were evaluated in both seasons (b). The determination coefficient describes the prediction accuracy across the two seasons
Considering a discard intensity of 20% of the hybrids with the lowest GV predicted in the winter crop season and comparing them with the same genotypes in the summer crop season, an error of only 10% was observed, which means that only eight promising genotypes would be erroneously discarded in the summer crop season upon taking the prediction data as a basis. In contrast, selection coincidence was only 41.3%, an index quite similar to the selection obtained from the 395 hybrids predicted in the winter and validated in the summer season (43%).
This set of hybrids predicted in the winter crop season and tested in the summer crop season was also used to verify if there are combinations between heterotic groups more predictable than others (Fig. 2).

Clustering of the lines within the groups of origin and correlation between the predicted GV in the winter crop season and the mean values observed in the summer crop season
The greatest correlations were observed in the crosses between the heterotic groups Amarillo Dent × Lancaster and Tropical Flint × Suwan. The lowest correlations were between the groups F-DK-Arg × Suwan, and Lancaster and NSSS crossed with Suwan. Negative correlations can also be observed in the crosses between Amarillo Dent × Tropical Flint and Tropical Dent × Suwan.
Discussion
Genetic parameters
In this study, the magnitude of the genetic variances in both crop seasons allows performing selection for grain yield in maize and to obtain moderate predictive ability for untested hybrids. Broad-sense heritabilities of the hybrids in the two crop seasons are in agreement with those observed in the literature (Windhausen et al. 2012). In the winter season, the magnitude of genotype-by-environment interaction (GEI) was higher than that in the summer one. Such result might prejudice the genomic predictions within the winter season and between both crop seasons (Lado et al. 2016). In the winter crop season, the additive and dominance variances showed very similar values, which highlight the importance of non-additive effects in maize grain yield. Technow et al. (2012) also observed an important contribution of dominant effects on the predictions in their dataset. Also, Guo et al. (2014) investigated the role of dominance effects in the heterosis and in the maize hybrid predictions. Their results showed the importance of dominance effect in the heterosis. This finding goes against the claim described in Troyer and Wellin (2009), who suggested that the additive participation in the heterosis has increased due to the improvement of lines. Nevertheless, it should be emphasized that some lines used in our study are derived from a temperate background and other ones from an introduced tropical background; that is, line origins ranged from Thai, Argentinean, and European to American materials, which may have contributed to an increase in dominance variance due to poor adaptation of some lines or because our crossing structure (given the number of backgrounds used) could be compared to complex pedigrees as observed in mapping studies.
In the summer crop season, the dominance variance was approximately three times greater than the additive variance, which shows the predominance of the non-additive effects acting in the grain yield trait, as observed in other studies using QTL mapping (Stuber et al. 1987; Guo et al. 2014). However, the predominance of dominance or over-dominance in maize is frequently found in QTL mapping studies, while in genome-wide selection (GWS), these effects have been reported as marginal, or as additional support for prediction (Technow et al. 2012; Santos et al. 2016).
Although in this study we do not evaluated the importance of dominance effect on the predictions (only its impact on the genotypic variance), in other simulation works, we observed that the inclusion of dominance effect can significantly improve the prediction ability (Santos et al. 2016).
The difference between DVs in the two experiments can also be explained by the favorable conditions found in the summer crop season. According to Hamblin and Morton (1977), dominance tends to be expressed more in favorable crop conditions. It should be noted that the average grain yield in the summer crop season was 8.21 t ha−1, while in the winter crop season, it was 5.33 t ha−1. This result emphasizes that the climatic conditions in the summer crop are superior to those observed in the winter crop.
The difference in the estimates of additive and dominance variances in the different crop seasons can also be explained by the different sets of lines used in each crop season. In the summer crop season, 374 lines were used, resulting in 794 single-cross hybrids. In the winter crop season, 300 lines were used, resulting in 838 hybrids. The two crop seasons had 262 lines in common, while 395 hybrids were in common which may also have caused the difference in the variance component estimates (Visscher et al. 2008).
Cross-validation and hybrid predictions
In genome prediction studies, the results of cross-validation have indicated that the GWS is more efficient than the classic marker-assisted selection methodology (Bernardo and Yu 2007) and that the GWS statistical models are very similar in their predictive abilities. Currently, one of the methods most used in GWS is GBLUP, which allows selection based on the genomic relationship information (Habier et al. 2007). The main difference between the statistical methods used in GWS is the assumptions about the distribution of the effects of the markers through the genome—more precisely, in the specification of the prior distributions of the marker effects. However, for quantitative traits as grain yield, which exhibit a polygenic or infinitesimal framework, these assumptions have not shown a significant influence on the correlations between predicted and observed values (Huang et al. 2010; Schön et al. 2004; Technow et al. 2014; Xua et al. 2014). In this context, given that the GBLUP method is computationally more efficient, it was chosen to be used in this study.
The predictive ability of the GBLUP model in the winter crop season was encouraging. The cross-validation results observed in our study were higher than those obtained in previous studies using the BLUP associated with low-density markers and identity-by-descent (Bernardo 1994, 1995, 1996a). Massman et al. (2013), using cross-validation and moderate density markers, obtained accuracies ranging from 0.75 to 0.87 for a level of 10% of missing hybrids using the rrBLUP model and phenotypic BLUP. Our results were very similar, considering the same level of missing hybrids in cross-validation (0.76 to 0.89). For the summer season, this correlation was slightly lower, varying from 0.56 to 0.73.
The ability of GBLUP to recovering the genetic variances estimated in the joint analysis shows the applicability of this method for recovering all the information available in the genome. Since the BLUP depends of accurate estimates of dispersion parameters, the more accurate the variance components are, the more accurate is the prediction ability. This claim becomes more evident when we compare the present results with the study developed by Cantelmo et al. (2016), which applied 79 microsatellite markers in only 51 lines of the same background from those used in this study and observed correlations ranging from 0.48 to 0.91. In other words, wide ranges of predictions were observed and the errors related to variance components were larger than those observed here.
In the study developed by Technow et al. (2014), using the GBLUP and Bayes B methodologies applied in a set of 1254 hybrids from the crossing between dent and flint heterotic groups evaluated in various years and locations and genotyped with approximately 35,000 SNP markers, observed cross-validation correlations ranged from 0.75 to 0.92 which also were similar to the results of our study. These authors used two heterotic groups (flint vs. dent), while in our work, we considered 13 backgrounds previously identified. In addition, we used some intra-group crosses; i.e., some crosses were realized within groups which allowed the prediction for all combinations including intra-group ones.
The intra-group crosses were built because doubts arose about the lines prior to classifications; i.e., some lines were clustered a priori as flint or dent, but their phenotypic characteristics and posterior marker genotype analysis indicated a different classification (see, for example, the tropical flint and tropical dent clustering in Fig. 2). It is evident that the cluster based on molecular markers is more generic than the classification based on ear kernel (Romay et al. 2013). These results suggest the importance of using all information provided by molecular markers to build the pedigree matrix and, in some cases, preferring the use of animal model over the partial diallel one, mainly when doubts arise about the line classification into heterotic groups.
The predictive accuracy for winter season did not decrease significantly as the rate training vs. validation population was reduced (Table 3). Results obtained in other studies show that with an increase in the size of the training population in proportion to the validation population, predictive ability also increases (Asoro et al. 2011; Technow et al. 2013). However, such scenario was observed only for summer season (Table 4). An explanation might be the high level of correlation already achieved in the missing level of 50% at winter season, as significant prediction increasing for low levels of missing hybrids was not possible (Technow et al. 2014).
One of the main factors involved in the accuracy of prediction of complex characteristics is the relation that exists between the training population and the validation population (Zhao et al. 2015). Gowda et al. (2013) reported a reduction of up to 93% in the prediction accuracy depending on the scenario of the relationship between the training population and validation population. Albrecht et al. (2011) also observed a decrease in the accuracy of prediction of general combining ability (GCA) using 1380 test crosses genotyped with 1152 SNP markers when the training and validation populations were not related. As a consequence of this dependency between accuracy of prediction and relationship between observed and predicted hybrids, the use of some models can at times generate negative correlations (Wang et al. 2014). For that reason, it is important that the training population be representative of the population that will be predicted so that the accuracy is truly representative of the predictive ability (Albrecht et al. 2011).
In another study, Albrecht et al. (2011), using the maize test cross found an increase in predictive accuracy when the amount of phenotypic data point was increased. In our study, the summer season was represented by four environments, and considering the natural unbalance that occurred across these locations, the length of the phenotypic vector was 3187 data points. On the other hand, the winter season was represented by six environments, and considering the missing natural data, the length of phenotypic vector was 5016 phenotypic data points. This difference in the amount of data may have affected the size of the correlations, especially for high levels of missing hybrids (i.e., 50% of missing data), where the main discrepancy between the two crop seasons was observed. It should be noted that the genotype-by-environment interaction was more evident in the winter crop season than in the summer crop season.
Since that in our case the length of dataset is related also to the number of environments, it could have influenced the prediction ability. In general, Burgueño et al. (2011) and Guo et al. (2013a) observed gains in predictive ability when several environments were used instead of only one location. Similar results could be observed in this study; that is, although the heritability of both crop seasons was similar, with 30% in the number of locations within the summer season compared to winter season, the correlation between predicted and observed GVs declined significantly.
These results were also observed by Albrecht et al. (2014); that is, the predictive ability may decrease with a decline in the number of locations. For grain yield, a reduction from 0.65 (with the set of four locations and 698 lines) to 0.32 (with one location and 87 lines) was observed. Similar results were observed by Burgueño et al. (2011), in which an increase in predictive ability occurred with an increase in the number of locations, regardless of environments highly correlated.
Influence of panel density on the predictions
The decrease in the density of the markers by half did not affect the predictive ability of the model. The stability of the correlations with the reduction in the number of markers can be observed, regardless the k-fold level adopted. This result corroborates that of Combs and Bernardo (2013), who verified that the gain in predictive accuracy reaches a threshold with a moderate number of markers in saturated genomes which depends on the population under study.
Albrecht et al. (2014), increasing the density of markers from 654 to 20,742 SNP markers for a set of 759 lines, observed an increase in predictive ability from 0.59 to 0.62. The same authors also observed that predictive ability could decrease with the use of a greater number of markers in the GBLUP model.
Wong and Bernardo (2008), using simulation data and the rrBLUP model in biparental populations, found that low density of markers could result in predictive abilities very near the maximum; that is, a decrease in the number of markers does not strongly affect the accuracy of prediction. These authors state that there is a maximum number of markers that, when exceeded, may result in a loss of accuracy. In another study, Jannink et al. (2010) decreased marker density by 75% and observed a decrease in prediction accuracy of only 0.03. Studies on dairy cattle have also shown similar results (Hayes et al., 2009a, b).
Simulation studies have suggested that, for certain models, the collinearity between linked markers can also decrease the accuracy of prediction (Zhong et al. 2009). This event may explain our results when the marker panel was decreased in 50% on the summer crop season and the value of correlation remained stable and even higher than those observed in full panel. Hayes et al. (2009a, b) suggest that the density of markers in genome-wide selection might be reduced if the training population presents some level of relationship with the validation population.
Prediction validation between crop seasons
In maize breeding programs, the genotypes are evaluated in multiple environment trials where their responses can be compared, the genotype-by-environment interaction are studied, and the best genotypes are selected across or within mega-environments (Crossa et al. 2010). Genomic information associated to phenotypic data can be used to predict the hybrid’s performance in different environments or years, reducing the cost related to phenotypic evaluation. However, in most studies with genomic prediction, the same dataset (or set of environments) is used to perform the predictions and cross-validation is applied to assess the GWS accuracy (Guo et al. 2013b). However, the effectiveness of the cross-validation methods is still highly questioned (Vitezica et al. 2013) and the results of this procedure are not always relevant to the breeder because the reality of a maize breeding program consists of evaluating genotypes in different environments and years. Thus, real prediction validation must be done across different locations and crop seasons to support the breeder decision in selecting the best genotypes.
In this scenario, we performed the validation, considering the different crop seasons and locations, and the efficiency of the GBLUP method in predicting hybrid’s performance was observed; in other words, the predictions were able to explain 28.4% of the variation of the hybrids in different crop seasons and locations. This result is quite relevant, given the heritability observed in both locations (0.66 and 0.67) and the influence of the genotype × environment interaction observed in the crop season used as the training population (winter season). Another explanation for this value is the correlation itself of the hybrids which were evaluated in both environments (0.61 or r 2 = 0.3603), showing the large influence of the effect of crop seasons and environments on prediction. Despite that, our result is very encouraging because the missing predictive ability was only 14%.
The correlation considering the crosses between the different heterotic groups was also verified. In this case, the most effective predictions were observed in the crosses belonging to different heterotic groups from different origins and/or within the same origins but combining materials of the flint vs. dent type. It was not mere coincidence that these combinations were the most exploited in the training population. The opposite also occurred; that is, highly negative correlations were observed in the combinations of heterotic groups less present in the training population and with combinations within the same origin and the same heterotic group. This result is very relevant since these misrepresented groups might present a low efficiency in the estimated of variance components if partial diallel models were used.
The results of the present study show that the magnitude of the correlations between predicted and observed hybrids both in the winter and summer crop seasons ranged from moderate to high, depending on the k-fold level in the cross-validation process. Also, a satisfactory ability of the GBLUP method to predict hybrids not tested in one crop season and their validation in different crop seasons was observed, even considering the moderate level of selective accuracy obtained among the hybrids evaluated in both seasons. In addition, it was found that an increase in the number of markers did not affect the predictive ability of the model.
It was concluded that the GBLUP method was able to generate high correlations between predicted and observed hybrids, even at high k-fold levels in the cross-validation process and in different locations and crop seasons.
References
Albrecht T et al (2011) Genome-based prediction of testcross values in maize. Theorical Applied Genetics 123:339–350
Albrecht T et al (2014) Genome-based prediction of maize hybrid performance across genetic groups, testers, locations, and years. Theorical Applied Genetics. 127:1375–1386
Asoro FG et al (2011) Accuracy and training population design for genomic selection on quantitative traits in elite North American oats. The Plant Genome 4(n. 2):132–144
Bernardo R (1995) Genetic models for predicting maize performance in unbalanced yield trial data. Crop Sci 35:141–147
Bernardo R (1996a) Best linear unbiased prediction of maize single cross performance. Crop Sci 36:50–56
Bernardo R (1996b) Best linear unbiased prediction of maize single-cross performance given erroneous inbred relationships. Crop Sci 36:862–866
Bernardo R (1994) Prediction of maize single-cross performance using RFLPs and information from related hybrids. Crop Sci 34:20–25
Bernardo R, Yu J (2007) Prospects for genomewide selection for quantitative traits in maize. Crop Sci 47:1082–1090
Burgueño J et al (2011) Prediction assessment of linear mixed models for multienvironment trials. Crop Scienc 51:944–954
Calus MPL (2010) Genomic breeding value prediction: methods and procedures. Animal 4:157–164
Cantelmo NF, Von Pinho RG, Balestre M (2016) Genomic breeding value prediction for simple maize hybrid yield using total effects of associated markers, under different imbalance levels and environments. Genet Mol Res 15. doi:10.4238/gmr.15017232
Cockerham CC (1954) An extension of the concept of partitioning hereditary variance for analysis of covariances among relatives when epistasis is present. Genetics 39:859–882
Combs E, Bernardo R (2013) Accuracy of genomewide selection for different traits with constant population size, heritability, and number of markers. The Plant Genome. 6:1–7
Crossa J et al (2010) Predictions of genetic values of quantitative traits in plant breeding using pedigree and molecular markers. Genetics 186:713–724
Crossa J et al (2013) Genomic prediction in maize breeding populations with genotyping-by-sequencing. G3 3:1903–1926
Endelman JB (2011) Ridge regression and other kernels for genomic selection with R package rrBLUP. The Plant Genome 4:250–255
Gowda M et al (2013) Best linear unbiased prediction of triticale hybrid performance. Euphytica 191:223–230
Guo T et al (2014) Genetic basis of grain yield heterosis in an “immortalized F2” maize population. Theor Appl Genet 127:2149–2158
Guo T et al (2013a) Performance prediction of F1 hybrids between recombinant inbred lines derived from two elite maize inbred lines. Theorical Applied Genetics. 126:189–201
Guo Z et al (2013b) Accuracy of across-environment genome-wide prediction in maize nested association mapping populations. G3 3:263–272
Habier D, Fernando RL, Dekkers JCM (2007) The impact of genetic relationship information on genome-assisted breeding values. Genetics 177:2389–2397
Hallauer AR, Carena M, Miranda JBF. (2010) Quantitative genetics in maize breeding. 3rd ed. Ames: Iowa State University.
Hamblin J, Morton JR (1977) Genetic interpretations of the effects of bulk breeding on four populations of beans (Phaseolus vulgaris L.) Euphytica 26:75–83
Hayes BJ et al (2009b) Invited review: genomic selection in dairy cattle: progress and challenges. J Dairy Sci 92:433–443
Hayes BJ, Visscher PM, Goddard ME (2009a) Increased accuracy of artificial selection by using the realized relationship matrix. Genet Res 91:47–60
Heslot N, Jannink JL, Sorrells ME (2014) Perspectives for genomic selection applications and research in plants. Crop Sci 55:1–12
Huang YF et al (2010) The genetic architecteture of grain yield and related traits in Zea maize L. revealed by comparing intermated and conventional populations. Genetics 186:395–404
Jannink JL, Lorenz AJ, Iwata H (2010) Genomic selection in plant breeding: from theory to practice. Briefings in Functional Genomics 9:166–177
Lado B, Barrios PG, Quincke M et al (2016) Modeling genotype by environment interaction for genomic selection with unbalanced data from a wheat breeding program. Crop Sci 56:2165–2179
Massman JM et al (2013) Genome wide predictions from maize single-cross data. Theorical Applied Genetics. 126:13–22
Meuwissen THE, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157:1819–1829
Nishio M, Satoh M (2014) Including dominance effects in the genomic BLUP method for genomic evaluation. PLoS One 9:1–6
Romay, M. C. et al. (2013). Comprehensive genotyping of the USA national maize inbred seed bank. Genome Biology, 1–18.
Santos JPR, Vasconcellos RCC, Pires LPM, Balestre M, Von Pinho RG (2016) Inclusion of dominance effects in the multivariate GBLUP model. PLoS One 11:e0152045
Schön CC et al (2004) Quantitative trait locus mapping based on resampling in a vast maize testcross experiment and its relevance to quantitative genetics for complex traits. Genetics 167:485–498
Schrag TA et al (2010) Prediction of hybrid performance in maize using molecular markers and joint analyses of hybrids and parental inbreds. Theorical Applied Genetics 120:451–461
Stuber CW, Edwards MD, Wendel JF (1987) Molecular marker-facilitated investigations of quantitative trait loci in maize. II. Factors influencing yield and its component traits. Crop Science 27:639–648
Technow F et al (2014) Genome properties and prospects of genomic prediction of hybrid performance in a breeding program of maize. Genetics 114:1–42
Technow F, Bürger A, Melchinger AE (2013) Genomic prediction of northern corn leaf blight resistance in maize with combined or separated training sets for heterotic groups. G3 3:197–203
Technow F, Riedelsheimer C, Schrag TA, Melchinger AE (2012) Genomic prediction of hybrid performance in maize with models incorporating dominance and population specific marker effects. Theor Appl Genet 125:1181–1194. doi:10.1007/s00122-012-1905-8
Troyer AF, Wellin EJ (2009) Heterosis decreasing in hybrids: yield test Inbreds. Crop Sci 49:1969–1976
VanRaden PM et al (2009) Invited review: reliability of genomic predictions for North American Holstein bulls. J Dairy Sci 92:16–24
VanRaden PM (2008) Efficient methods to compute genomic predictions. J Dairy Sci 91:4414–4423
Visscher PM, Hill WG, Wray NR (2008) Heritability in the genomics era—concepts and misconceptions. Nat Rev Genet 91:255–266
Vitezica ZG, Varona L, Legarra A (2013) On the additive and dominant variance and covariance of individuals within the genomic selection scope. Genetics 195:1223–1230
Wang Y et al (2014) The accuracy of prediction of genomic selection in elite hybrid rye populations surpasses the accuracy of marker-assisted selection and is equally augmented by multiple field evaluation locations and test years. BMC Genomics 15:556
Windhausen VS et al (2012) Effectiveness of genomic prediction of maize hybrid performance in different breeding populations and. Environments G3 2:1427–1436
Wong C, Bernardo R (2008) Genomewide selection in oil palm: increasing selection gain per unit time and cost with small populations. Theorical Applied Genetics, Berlin 116:815–824
Xua S, Zhub D, Zhang Q (2014) Predicting hybrid performance in rice using genomic best linear unbiased prediction. PNAS 111:12456–12461
Zeng ZB, Wang T, Zou W (2005) Modeling quantitative trait loci and interpretation of models. Genetics 169:1711–1725
Zhao Y, Mette MF, Reif JC (2015) Genomic selection in hybrid breeding. Plant Breed 134:1–10
Zhong S et al (2009) Factors affecting accuracy from genomic selection in populations derived from multiple inbred lines: a barley case study. Genetics 182:355–364
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Cantelmo, N.F., Von Pinho, R.G. & Balestre, M. Genome-wide prediction for maize single-cross hybrids using the GBLUP model and validation in different crop seasons. Mol Breeding 37, 51 (2017). https://doi.org/10.1007/s11032-017-0651-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-017-0651-7