
Abstract
Tobacco (Nicotiana tabacum L., 2n = 48) is an important agronomic crop and model plant. Flue-cured tobacco is the most important type and accounts for approximately 80 % of tobacco production worldwide. The low genetic diversity of flue-cured tobacco impedes the construction of a high-density genetic linkage map using simple sequence repeat (SSR) markers and warrants the exploitation of single nucleotide polymorphic (SNP) markers from genomic regions. In this article, initially using specific locus-amplified fragment sequencing, we discovered 10,891 SNPs that were subsequently used as molecular markers for genetic map construction. Combined with SSR markers, a final high-density genetic map was generated containing 4215 SNPs and 194 SSRs distributed on 24 linkage groups (LGs). The genetic map was 2662.43 cM in length, with an average distance of 0.60 cM between adjacent markers. Furthermore, by mapping the SNP markers to the ancestral genomes of Nicotiana tomentosiformis and Nicotiana sylvestris, a large number of genome rearrangements were identified as occurring after the polyploidization event. Finally, using this novel integrated map and mapping population, two major quantitative trait loci (QTLs) were identified for flue-curing and mapped to the LG6 of tobacco. This is the first report of SNP markers and a SNP-based linkage map being developed in tobacco. The high-density genetic map and QTLs related to tobacco curing will support gene/QTL fine mapping, genome sequence assembly and molecular breeding in tobacco.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Common tobacco (Nicotiana tabacum L.) is an important non-food crop grown worldwide that also serves as a model organism. Tobacco is an allotetraploid (2n = 4x = 48) belonging to the Solanaceae family and is most likely the result of a tetraploidization event involving Nicotiana sylvestris (S-genome) and Nicotiana tomentosiformis (T-genome) (Lim et al. 2004; Clarkson et al. 2005). Substantial and rapid research progress in Nicotiana genomics has been made in recent years (Wang and Bennetzen 2015). Multiple genetic maps have been constructed for tobacco to better understand its genome and assist in breeding efforts, among other applications. Targeting unmethylated gene-rich regions, more than 1.3 million genome survey sequences were produced by the Tobacco Genome Initiative (Gadani et al. 2003). Based on these sequences, a simple sequence repeat (SSR)-based linkage map was generated using an F2 population derived from two different parental tobacco lines (‘Hicks Broad Leaf’ × ‘Red Russian’), containing 2317 microsatellite markers and 2363 loci (Bindler et al. 2007, 2011). A linkage map of flue-cured tobacco was constructed based on a double haploid population from a cross between ‘HHDJY’ and ‘Hicks Broad Leaf’ using 851 markers, including SSRs, and diversity array technology (DArT) (Lu et al. 2013). As observed in many other crops, stringent quality requirements in breeding selection have resulted in a reduction in tobacco genetic diversity over recent decades (Moon et al. 2009a, b). An extremely narrow genetic diversity in the flue-cured tobacco pool was revealed by several molecular marker-based studies (Zhang et al. 2006; Yang et al. 2007). Single-nucleotide polymorphisms (SNPs) have provided abundant variability for developing markers in many crop species, including maize (Ching et al. 2002) and soybean (Zhu et al. 2003).
The rapid development of next-generation sequencing technologies has made it possible to complete the genome sequencing of many larger crop species (Tomato Genome Consortium 2012; Potato Genome Sequencing et al. 2011; Kim et al. 2014; Wang et al. 2012a; Brenchley et al. 2012), including four allotetraploid Nicotiana (N. benthamiana plus three cultivars of N. tabacum: TN90, K326 and Basma Xanthi) and three diploid Nicotiana (N. otophora, N. sylvestris and N. tomentosiformis) (Bombarely et al. 2012; Sierro et al. 2014, 2013a). The genome sizes were estimated to be 4.5 Gb for N. tabacum. Approximately 3.7 Gb of assembled sequences is available for each variety in GenBank. However, for large genomes and populations, whole-genome resequencing remains cost-prohibitive. Hence, efficient methods for large-scale genotyping based on a reduced representation library combined with high-throughput sequencing, restriction site-associated DNA sequencing (Miller et al. 2007), two-enzyme genotyping by sequencing (Poland et al. 2012) and specific-locus amplified fragment sequencing (SLAF-seq) (Sun et al. 2013) have been developed. Many high-density genetic maps have been constructed using these methods in species such as pear (Wu et al. 2013), eggplant (Barchi et al. 2012), ryegrass (Pfender et al. 2011), grape (Wang et al. 2012b) and sesame (Zhang et al. 2013). As for flue-cured tobacco, due to the large genome size, SLAF-seq is an appropriate high-resolution strategy for the large-scale SNP discovery and genotyping required to create a genetic map.
In this study, we employed SLAF-seq to achieve the first rapid mass discovery of SNP markers and high-density genetic mapping in flue-cured tobacco. Additionally, a comparative genome analysis and the identification of curing-related quantitative trait loci (QTLs) based on these markers and the resulting genetic map are presented.
Materials and methods
Plant materials and DNA extraction
A population of 100 F2 plants derived from “Yunyan 85” × “Dabaijin 599” was used to construct the genetic map and detect curing-related QTLs. The breeding lines were different from one another in curing traits. “Yunyan 85” is easy to cure, while “Dabaijin 599” is difficult to cure. Young leaves were collected and frozen in liquid nitrogen for DNA extraction. Total genomic DNA was extracted according to the cetyltrimethylammonium bromide method. DNA concentrations and qualities were estimated using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Milan, Italy) and electrophoresis in agarose gels.
Marker data generation
The mapping population was genotyped with SSR and SNP markers. In total, 4710 SSR markers for tobacco were used, with 2363 being reported previously (Bindler et al. 2011), and 2347 SSR markers were developed with MISA software using the tobacco genome (Thiel et al. 2003). Repeat units of 2–6 nucleotides were considered. The minimum number of repeats for SSR detection was defined as six for di-SSRs, five for tri-SSRs, four for tetra-SSRs, three for penta-SSRs and three for hexa-SSRs. The sequence of the tobacco genome assembly was downloaded from GenBank (AWOJ00000000). Polymerase chain reactions (PCRs) were performed in 10-μl reactions, containing 25 ng DNA, 2 pmol of each primer, 2 nmol dNTPs, 20 nmol MgCl2, 2U Taq DNA polymerase and 1 × PCR buffer supplied together with the enzyme. The PCR cycles were 94 °C for 5 min, 33 cycles of 94 °C for 15 s, 55–62 °C (depending on the primers) for 15 s, 72 °C for 30 s and 7 min at 72 °C for the final extension. PCR products were separated in 8 % polyacrylamide gels and were visualized by silver staining.
SLAF-seq was used for the rapid and effective discovery of SNP markers. The SLAF library was constructed as described by Sun et al. (2013), with small modifications. The digestion sites and the length and distribution of the resulting fragments in the reference genome were investigated, and the genomic DNA of the two parents and the F2 population was digested using the restriction enzymes HaeIII and BfaI. Each DNA sample was assigned a unique barcode. The fragments of 330–380 bp were selected to generate paired-end reads on an Illumina HiSeq-2000 sequencing platform (Illumina, San Diego, CA, USA). Then low-quality reads (quality score ≤ Q20) were filtered out and barcode sequences were trimmed. Finally, we selected 45-bp paired reads to develop SLAF loci. All of the reads were mapped onto the tobacco genome sequence using SOAP2 software (Li et al. 2009). SOAP2 is an improved ultrafast tool for short read alignments. Additionally, SOAP2 is compatible with both single- and paired-end reads. Sequences mapping to the same position defined an SLAF locus if the polymorphism between the parents was identified. SLAF markers were filtered and assessed for quality using the methods described by Sun et al. (2013). A high-quality SLAF marker contains <3 SNPs, and the average depth of each sample is >3. Markers having full parental homozygosity were used to construct the genetic map.
Linkage map construction
The HighMap method was used to order the SNP and SSR markers and correct genotyping errors (Liu et al. 2014). The MSTmap algorithm was used to order markers (Wu et al. 2008), and the SMOOTH algorithm (van Os et al. 2005) was used to correct genotyping errors following marker ordering. The processes of ordering and error correction were conducted iteratively to produce an accurate map order and distance. Finally, heat and haplotype maps were constructed to evaluate the map quality (West et al. 2006). Map distances were estimated using the Kosambi mapping function (Kosambi 1944).
Ancestral origin assignment
The sequences of map-assigned SNPs were used to perform alignment with the genome assemblies of the two diploid ancestors N. tomentosiformis and N. sylvestris (GenBank BioProjects no: PRJNA182500 and PRJNA182501). The SOAP2 program (Li et al. 2009) was used to conduct similarity searches.
Trait evaluation and QTL analysis
In total, 100 F2 plants from “Yunyan 85” × “Dabaijin 599,” as well as both of the parental lines, were grown under normal greenhouse conditions in 2011. Additionally, 100 F2:3 families obtained by selfing each F2 individual were grown on the Jimo Experiment Farm in 2012 and 2013. The tobacco seedlings were transplanted into cell trays 3 weeks after the beginning of germination. One month after transplantation, the tobacco plants were transferred to the field. After 3 months, the tops can be removed before any flowers have opened, and the plant retains 20 leaves. Then, axillary buds or suckers were removed. The 11th and 12th leaves were cut approximately 1 month after topping. The curing traits were measured using the leaf yellowing percentage (LY) in a dark thermostatic-humidistatic cultivating box with an air temperature of 36 °C and relative humidity of 90 %. The demarcation of the border between yellow and green was obvious. The “yellowing percentage area” can be calculated using the histogram window in Photoshop software after treatments of 24, 48, 72 and 96 h. The higher mean values represent easy curing. Meanwhile, the LY indices for each F2:3 generation were scored as the mean values of ten progeny.
MapQTL 5.0 was used to map the QTLs using the interval mapping method (Ooijen 2004). A QTL was declared as significant when its logarithm of odds (LOD) value exceeded the threshold of 3. The maximum LOD score in the interval was taken as the position of the QTL. The confidence intervals were established as the map intervals corresponding to 1 LOD on either side of the LOD peak. Additive and dominant genetic effects, as well as the proportion of the variance explained by each QTL (PVE), were estimated.
Results
SSR and SNP discovery
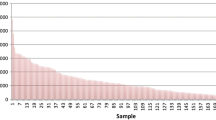
A total of 2347 novel markers were developed under the minimal stringency conditions, and 77 SSRs (3.3 %) had polymorphic band patterns between the parents of the mapping population, which were used for additional analysis. In total, 160 (6.8 %) of 2363 known SSR markers were polymorphic between the parents. Subsequently, a large number of SNP markers was discovered by means of SLAF-seq. After high-throughput sequencing and preprocessing, 223,005,697 high-quality paired-end reads were obtained, with each read being ~45 bp in length. Of these, 31,459,177 reads were from the male parent, and 16,205,350 reads were from the female parent, with the remaining being from 100 individuals of the F2 population. Read numbers for the individuals ranged from 667,049 to 4,924,655, with an average of 1,719,887. The supporting data are available in the NCBI SRA repository (PRJNA269005). Furthermore, 379,006 SLAFs were detected by group, and the numbers of SLAFs in the male and female parents were 355,343 and 265,414, respectively. For the F2 population, the number of SLAFs in each individual ranged from 147,650 to 304,268 with an average of 218,907 (Fig. 1). The average sequencing depth of the SLAFs was 51.91 in the male parent, 33.33 in the female parent, and 4.23 in the progeny (Fig. 2). Among these SLAFs, 21,959 were polymorphic, indicating a polymorphism rate of only 5.79 %. After filtering out the SLAFs that lacked parental information, 14,252 could be classified into eight segregation patterns, and ultimately 10,891 high-quality markers had parental homozygosity with over 70 % integrity and were chosen to construct a linkage map.

Number of SLAFs in each progeny. The x-axis represents individual F2 plant accessions, and the y-axis represents the number of specific locus-amplified fragments (SLAFs)

Sequencing depth of SLAFs and markers in each progeny. The x-axis represents individual F2 plant accessions, and the y-axis represents the average sequencing depth of the specific locus-amplified fragments (SLAFs) and markers
Genetic mapping
A total of 11,125 markers (10,891 SNPs and 237 SSRs) were used to construct a high-density genetic map. Finally, 4409 markers (4215 SNPs and 194 SSRs) were mapped to 24 linkage groups (LGs) (Fig. 3; Additional files 1, 2 and 3). However, 38.58 % of the markers cosegregate, and the genetic map contains 2781 loci. The average sequencing depths of these markers were 78.86 × in ‘Yunyan 85,’ 40.08 × in ‘Dabaijin 599’ and 5.04 × in each F2 individual (Fig. 2). The number of LGs corresponded to the number of chromosomes (x = 24). The final map covered 2662.43 cM, with an average distance of 0.60 cM between adjacent markers (Table 1). Each LG contained an average of 184 markers covering an average of 110.93 cM. The genetic length of the LGs ranged from 26.35 to 273.25 cM. The largest LG (LG2) included 288 markers spanning 273.25 cM, with an average intermarker distance of 0.95 cM. The smallest LG (LG7) contained 95 markers covering a length of 26.35 cM, with an average intermarker distance of 0.91 cM. LG2 and LG20 had the highest average marker density (0.17 cM and 0.19 cM), whereas LG10 and LG14 had the greatest average intermarker distance (1.47 cM). The degree of linkage between markers was reflected by “Gaps ≤ 5,” which ranged from 97.50 to 100.00 %, with an average value of 99.27 %.


High-density genetic map of the flue-cured tobacco genome. Co-segregating markers (± 0.5 cM) were collapsed into unique loci with a single representative marker. Red markers are assigned to the S-genome. Blue markers are assigned to the T-genome. Marker names are shown to the right of each chromosome, with map distances (in cM) shown on the left
The haplotype map reflects genotyping errors caused by double crossovers and the recombination events of each individual (Additional file 4). Most of the recombination blocks were exactly defined. The heat map reflects the relationship of the recombination between markers and is used to find ordering errors and evaluate map quality (Additional file 5). Most of the LGs performed well based on these visualizations.
Ancestral origin assignment
According to the alignment, 3900 (92 %) markers could be mapped to the assembly of the flue-cured tobacco ‘K326’ (Sierro et al. 2014). Furthermore, the SNP markers were assigned to the two diploid ancestral genomes of common tobacco. Of all of the markers, 49.7 % were mapped to the S-genome and 38.7 % to the T-genome. Those remaining could not be assigned because of sequence diversity or poor genome coverage. The genetic distance covered on the S-genome with 2095 markers was longer than that covered on the T-genome with 1633 markers (Table 1). For most of the LGs, the markers were predominantly derived from one of the two genomes. Seventeen LGs could be clearly assigned to either the S- or T-genome. Seven LGs contained both S- and T-genome-specific markers, most of which were grouped in different parts of the LGs (Fig. 3). For example, on LG1, the top half of the LG was derived from the S-genome and the rest was derived from the T-genome.
QTL analysis
The two parents differed markedly (P < 0.01), and the progeny displayed a continuous distribution, in the aspect of curing. The LY was 30 % for ‘Dabaijin 599’ and 55 % for ‘Yunyan 85.’ The correlation of LY to different years was strong, with coefficients >0.65. Based on the high-density genetic map, the QTLs associated with leaf curing in the F2 population over 1 year and in the F2:3 over 2 years were analyzed separately. Seven QTLs on LG6 and one QTL on LG4 were detected at a LOD score >3 (Table 2). Among them, two major QTLs, 2011QLY-6-1 and 2011QLF-6-2, on LG6 were reliably detected in the F2 and F2:3 populations over 3 years. 2011QLY-6-1 explained 19.3 % of the total variation of leaf LY and was located at 58.7 cM based on the peak marker Marker23312 on LG6, with an LOD value of 4.36. QTL 2011QLF-6-2 explained 15.2 % of the total variation of LY and was located at 97.6 cM on the same chromosome, with an LOD value of 3.59 and the peak marker Marker42089. 2012QLY-6-1 and 2013QLY-6-1 were located in the same region as 2011QLY-6-1 on LG6 and explained higher percentages of variation. 2012QLY-6-2, 2013QLY-6-2, and 2011QLY-6-2 were found to be located in the same chromosomal region. One major QTL, 2011QLY-4-1, was only found in the F2 population and explained 13.8 % of the total LY variation.
Discussion
Construction of a high-resolution genetic map based on the SLAF-seq
A high-density linkage map could bridge the gap between genomic and targeted breeding. The level of polymorphisms presented in cultivated flue-cured tobacco is low, as in many other crop species. A total of 2415 (47 %) functional markers were polymorphic between the parent ‘Hicks Broadleaf’ (a flue-cured tobacco type) and ‘Red Russian’ populations (specialty tobacco) (Bindler et al. 2011). In contrast, there is only a 4.9 % polymorphism rate for SSR markers between the flue-cured tobacco parents of the mapping population, indicating that an extremely narrow genetic diversity range impedes the construction of a high-density genetic linkage map using SSR markers. Fortunately, current high-throughput sequencing technologies combined with SLAF have facilitated the rapid discovery of a large number of SNPs in flue-cured tobacco. We discovered more than 21,959 SNPs from ~380 K SLAF tags. The polymorphism rate of SLAFs was 5.78 %, and one SNP was observed per 1.5 kbp across the genome. These results accurately reflect the genetic and polymorphic characteristics of flue-cured tobacco and the great advantage of SNP markers, which have a much higher frequency than SSRs (Bindler et al. 2011). SNPs are more useful as genetic markers than SSRs because they are stable in most genomes and their detection is adaptable to high-throughput automation. A total of 10,891 high-quality polymorphic SNP markers were identified for genetic linkage map construction. Thus, SLAF-seq is suitable for the large-scale development of molecular markers, having a high success rate, specificity, and stability at a low cost.
Furthermore, 4215 novel developed SNPs were assigned to 24 LGs corresponding to the 24 chromosome pairs of the tobacco genome. As an allotetraploid species, polyploidization makes the identification of SNPs more complicated. The large number of homoeologous and paralogous sequences in the genomes resulted in the discovery of many useless non-allelic SNPs. Moreover, it was difficult to obtain accurate genotyping based on short reads because of the low genome coverage and sequencing errors. Thus, many polymorphic markers could not be mapped and were eliminated from the mapping experiment. The entire genetic length of the map was 2662 cM, slightly shorter than that obtained by Bindler et al. (2011). However, the length was substantially longer than that of the DArT-based flue-cured tobacco map (Lu et al. 2013). Compared with previously published genetic maps of tobacco, the map in the present study has a higher marker density, with a mean intermarker distance of 0.60 cM. The marker density was nonuniform because of sequence depth, reference genome quality and restriction enzymes. Meanwhile, most of the chromosomes indicated a marker cluster, which is usually associated with the centromere and its constitutive heterochromatin (Tanksley et al. 1992). In this work, the abundant number of SNP markers and the small plant populations caused the map to be saturated, and 38.58 % of the markers cosegregated. A larger mapping population could increase the resolution of the map. In further research, we will increase the population size to finely map the associated QTLs.
Applications of the genetic map
Next-generation sequencing technology has enabled the rapid generation of draft genomes for tobacco (Sierro et al. 2013a, 2014). Nevertheless, the large number of repetitive DNA sequences in tobacco has hindered assembly accuracy and length. Consequently, a high-quality genetic map is an appropriate way to anchor scaffolds onto chromosomes. In this study, 3900 (92 %) SNP markers were mapped to the de novo assembly of the flue-cured tobacco ‘K326.’ However, these scaffold sequence lengths were too short; thus, most of the scaffolds could not be anchored to the genetic map. More effort into the genome sequencing of tobacco is necessary to anchor and orient the scaffolds on the chromosomes using the high-resolution SNP genetic map of this species. By mapping SNP markers to the sequenced ancestral genomes of N. tomentosiformis and N. sylvestris, corresponding regions of the N. tabacum genome with different ancestral origins have been identified. Seven LGs contained both S- and T-genome-specific markers, indicating that the two genomes were rearranged after the polyploidization event, which was similar to the result of Bindler et al. (2011). Based on the SSR amplification in N. sylvestris and N. tomentosiformis, the four linkage groups consisted of both S- and T-genome-specific markers mainly grouped in different sections of their respective linkage groups (Bindler et al. 2011). Moreover, the sequence identity with diploid donor species and the WGP physical map indicated that a number of chromosomal loci rearrangements occurred in every linkage group and in the inversion of the S and T assignments in linkage group 22 (Sierro et al. 2013b, 2014). Our analysis confirms the rearrangements in most linkage groups. This result is also supported by cytogenetic analyses (Lim et al. 2007) and COSII genetic maps (Wu et al. 2010). Additionally, genome deletions may have occurred since the speciation event (Lim et al. 2004; Doyle et al. 2008; Renny-Byfield et al. 2011; Sierro et al. 2014).
Base on this high-density linkage map, we first reported QTL influencing a flue-curing trait. LY is an important index for evaluating curing traits. The average phenotypic values of the F2:3 progeny derived from selfing F2 plants, and the F2 phenotype itself, were used in this study. A QTL is only classified as "major" if it accounts for >10 % of the PVE (Collard et al. 2005). In this study, three major QTLs in the F2 plants and four major QTLs in the F2:3 progeny were detected. Furthermore, major QTLs must be stable across multiple seasons/locations (Li et al. 2001; Pilet-Nayel et al. 2002). The two major QTLs on LG6 were stably identified over 3 years, which together explained ~50 % of the phenotypic variance for LY. These stable QTLs are the most promising for the marker-assisted selection of curing traits in tobacco.
Conclusions
Despite the low level of genetic variation present in flue-cured tobacco, a large number of SNP markers using SLAF-seq technology were generated, and a high-density SNP genetic map was successfully constructed. This genetic map is the densest map to date for tobacco. Complex genomic rearrangements were identified using comparative genomic analyses with the two diploid ancestral genomes. In addition, two repeatable major QTLs for curing traits were located, and these loci together explained the majority of the phenotypic variance. These results confirmed the reliability of the genetic map for future gene/QTL fine mapping, sequence assembly, comparative genomic analysis and molecular breeding in tobacco.
References
Barchi L, Lanteri S, Portis E, Vale G, Volante A, Pulcini L, Ciriaci T, Acciarri N, Barbierato V, Toppino L, Rotino GL (2012) A RAD tag derived marker based eggplant linkage map and the location of QTLs determining anthocyanin pigmentation. PLoS One 7(8):e43740. doi:10.1371/journal.pone.0043740
Bindler G, van der Hoeven R, Gunduz I, Plieske J, Ganal M, Rossi L, Gadani F, Donini P (2007) A microsatellite marker based linkage map of tobacco. Theor Appl Genet 114(2):341–349
Bindler G, Plieske J, Bakaher N, Gunduz I, Ivanov N, Van der Hoeven R, Ganal M, Donini P (2011) A high density genetic map of tobacco (Nicotiana tabacum L.) obtained from large scale microsatellite marker development. Theor Appl Genet 123(2):219–230
Bombarely A, Rosli HG, Vrebalov J, Moffett P, Mueller L, Martin G (2012) A draft genome sequence of Nicotiana benthamiana to enhance molecular plant-microbe biology research. Mol Plant Microbe Interact 25:1523–1530
Brenchley R, Spannagl M, Pfeifer M et al (2012) Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 491(7426):705–710. doi:10.1038/nature11650
Ching A, Caldwell KS, Jung M, Dolan M, Smith OS, Tingey S, Morgante M, Rafalski AJ (2002) SNP frequency, haplotype structure and linkage disequilibrium in elite maize inbred lines. BMC Genet. doi:10.1186/1471-2156-3-19
Clarkson JJ, Lim KY, Kovarik A, Chase MW, Knapp S, Leitch AR (2005) Long-term genome diploidization in allopolyploid Nicotiana section Repandae (Solanaceae). New Phytol 168(1):241–252. doi:10.1111/j.1469-8137.2005.01480.x
Collard BCY, Jahufer MZZ, Brouwer JB, Pang ECK (2005) An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: the basic concepts. Euphytica 142(1–2):169–196. doi:10.1007/s10681-005-1681-5
Doyle JJ, Flagel LE, Paterson AH, Rapp RA, Soltis DE, Soltis PS, Wendel JF (2008) Evolutionary genetics of genome merger and doubling in plants. Annu Rev Genet 42:443–461. doi:10.1146/annurev.genet.42.110807.091524
Gadani F, Hayes A, Opperman CH et al (2003) Large scale genome sequencing and analysis of Nicotiana tabacum: the tobacco genome initiative. In: Proceedings, 5èmes Journées ScientiWques du Tabac de Bergerac—5th bergerac tobacco ScientiWc meeting, Bergerac, pp 117–130. http://www.altadis-bergerac.com/pdf/5_JS_Bergerac.pdf
Kim S, Park M, Yeom SI et al (2014) Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species. Nat Genet 46(3):270–278. doi:10.1038/ng.2877
Kosambi DD (1944) The estimation of map distance from recombination values. Ann Eugen 12:172–175
Li Z, Jakkula L, Hussey RS, Tamulonis JP, Boerma HR (2001) SSR mapping and confirmation of the QTL from PI96354 conditioning soybean resistance to southern root-knot nematode. Theor Appl Genet 103(8):1167–1173. doi:10.1007/s001220100672
Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J (2009) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25(15):1966–1967. doi:10.1093/bioinformatics/btp336
Lim KY, Matyasek R, Kovarik A, Leitch AR (2004) Genome evolution in allotetraploid Nicotiana. Biol J Linn Soc 82(4):599–606. doi:10.1111/j.1095-8312.2004.00344.x
Lim KY, Kovarik A, Matyasek R, Chase MW, Clarkson JJ, Grandbastien MA, Leitch AR (2007) Sequence of events leading to near-complete genome turnover in allopolyploid Nicotiana within five million years. New Phytol 175(4):756–763. doi:10.1111/j.1469-8137.2007.02121.x
Liu D, Ma C, Hong W, Huang L, Liu M, Liu H, Zeng H, Deng D, Xin H, Song J, Xu C, Sun X, Hou X, Wang X, Zheng H (2014) Construction and analysis of high-density linkage map using high-throughput sequencing data. PLoS One 9(6):e98855. doi:10.1371/journal.pone.0098855
Lu XP, Gui YJ, Xiao BG, Li YP, Tong ZJ, Liu Y, Bai XF, Wu WR, Xia L, Huttner E, Kilian A, Fan LJ (2013) Development of DArT markers for a linkage map of flue-cured tobacco. Chin Sci Bull 58(6):641–648. doi:10.1007/s11434-012-5453-z
Miller MR, Dunham JP, Amores A, Cresko WA, Johnson EA (2007) Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res 17(2):240–248. doi:10.1101/Gr.5681207
Moon HS, Nicholson JS, Heineman A, Lion K, van der Hoeven P, Hayes AJ, Lewis RS (2009a) Changes in genetic diversity of US flue-cured tobacco germplasm over seven decades of cultivar development. Crop Sci 49(2):498–508. doi:10.2135/cropsci2008.05.0253
Moon HS, Nifong JM, Nicholson JS, Heineman A, Lion K, van der Hoeven R, Hayes AJ, Lewis RS (2009b) Microsatellite-based analysis of tobacco (Nicotiana tabacum L.) genetic resources. Crop Sci 49(6):2149–2159. doi:10.2135/cropsci2009.01.0024
Ooijen V (2004) MapQTL® 5, software for the mapping of quantitative trait loci in experimental populations. Kyazma BV, Wageningen
Pfender WF, Saha MC, Johnson EA, Slabaugh MB (2011) Mapping with RAD (restriction-site associated DNA) markers to rapidly identify QTL for stem rust resistance in Lolium perenne. Theor Appl Genet 122(8):1467–1480. doi:10.1007/s00122-011-1546-3
Pilet-Nayel ML, Muehlbauer FJ, McGee RJ, Kraft JM, Baranger A, Coyne CJ (2002) Quantitative trait loci for partial resistance to Aphanomyces root rot in pea. Theor Appl Genet 106(1):28–39. doi:10.1007/s00122-002-0985-2
Poland JA, Brown PJ, Sorrells ME, Jannink JL (2012) Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS One. doi:10.1371/journal.pone.0032253
Potato Genome Sequencing C, Xu X, Pan S et al (2011) Genome sequence and analysis of the tuber crop potato. Nature 475(7355):189–195. doi:10.1038/nature10158
Renny-Byfield S, Chester M, Kovarik A, Le Comber SC, Grandbastien MA, Deloger M, Nichols RA, Macas J, Novak P, Chase MW, Leitch AR (2011) Next generation sequencing reveals genome downsizing in allotetraploid Nicotiana tabacum, predominantly through the elimination of paternally derived repetitive DNAs. Mol Biol Evol 28(10):2843–2854. doi:10.1093/molbev/msr112
Sierro N, Battey JN, Ouadi S, Bovet L, Goepfert S, Bakaher N, Peitsch MC, Ivanov NV (2013a) Reference genomes and transcriptomes of Nicotiana sylvestris and Nicotiana tomentosiformis. Genome Biol 14(6):R60. doi:10.1186/gb-2013-14-6-r60
Sierro N, van Oeveren J, van Eijk MJ, Martin F, Stormo KE, Peitsch MC, Ivanov NV (2013b) Whole genome profiling physical map and ancestral annotation of tobacco Hicks Broadleaf. Plant J 75(5):880–889
Sierro N, Battey JN, Ouadi S, Bakaher N, Bovet L, Willig A, Goepfert S, Peitsch MC, Ivanov NV (2014) The tobacco genome sequence and its comparison with those of tomato and potato. Nat Commun 5:3833. doi:10.1038/ncomms4833
Sun XW, Liu DY, Zhang XF et al (2013) SLAF-seq: an efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS One. doi:10.1371/journal.pone.0058700
Tanksley SD, Ganal MW, Prince JP, de Vicente MC, Bonierbale MW, Broun P, Fulton TM, Giovannoni JJ, Grandillo S, Martin GB et al (1992) High density molecular linkage maps of the tomato and potato genomes. Genetics 132(4):1141–1160
Thiel T, Michalek W, Varshney RK, Graner A (2003) Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet 106(3):411–422. doi:10.1007/s00122-002-1031-0
Tomato Genome Consortium (2012) The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485(7400):635–641. doi:10.1038/nature11119
van Os H, Stam P, Visser RGF, van Eck HJ (2005) SMOOTH: a statistical method for successful removal of genotyping errors from high-density genetic linkage data. Theor Appl Genet 112(1):187–194. doi:10.1007/s00122-005-0124-y
Wang X, Bennetzen JL (2015) Current status and prospects for the study of Nicotiana genomics, genetics, and nicotine biosynthesis genes. Mol Genet Genomics 290(1):11–21. doi:10.1007/s00438-015-0989-7
Wang K, Wang Z, Li F et al (2012a) The draft genome of a diploid cotton Gossypium raimondii. Nat Genet 44(10):1098–1103. doi:10.1038/ng.2371
Wang N, Fang LC, Xin HP, Wang LJ, Li SH (2012b) Construction of a high-density genetic map for grape using next generation restriction-site associated DNA sequencing. BMC Plant Biol 12:148. doi:10.1186/1471-2229-12-148
West MA, van Leeuwen H, Kozik A, Kliebenstein DJ, Doerge RW, St Clair DA, Michelmore RW (2006) High-density haplotyping with microarray-based expression and single feature polymorphism markers in Arabidopsis. Genome Res 16(6):787–795. doi:10.1101/gr.5011206
Wu YH, Bhat PR, Close TJ, Lonardi S (2008) Efficient and accurate construction of genetic linkage maps from the minimum spanning tree of a graph. PLoS Genet. doi:10.1371/journal.pgen.1000212
Wu FN, Eannetta NT, Xu YM, Plieske J, Ganal M, Pozzi C, Bakaher N, Tanksley SD (2010) COSII genetic maps of two diploid Nicotiana species provide a detailed picture of synteny with tomato and insights into chromosome evolution in tetraploid N. tabacum. Theor Appl Genet 120(4):809–827. doi:10.1007/s00122-009-1206-z
Wu J, Wang Z, Shi Z et al (2013) The genome of the pear (Pyrus bretschneideri Rehd.). Genome Res 23(2):396–408. doi:10.1101/gr.144311.112
Yang BC, Xiao BG, Chen XJ, Shi CH (2007) Assessing the genetic diversity of tobacco germplasm using intersimple sequence repeat and inter-retrotransposon amplification polymorphism markers. Ann Appl Biol 150(3):393–401. doi:10.1111/j.1744-7348.2007.00139.x
Zhang HY, Liu XZ, Li TS, Yang YM (2006) Genetic diversity among flue-cured tobacco (Nicotiana tabacum L.) revealed by amplified fragment length polymorphism. Bot Stud 47(3):223–229
Zhang Y, Wang L, Xin H, Li D, Ma C, Ding X, Hong W, Zhang X (2013) Construction of a high-density genetic map for sesame based on large scale marker development by specific length amplified fragment (SLAF) sequencing. BMC Plant Biol 13:141. doi:10.1186/1471-2229-13-141
Zhu YL, Song QJ, Hyten DL, Van Tassell CP, Matukumalli LK, Grimm DR, Hyatt SM, Fickus EW, Young ND, Cregan PB (2003) Single-nucleotide polymorphisms in soybean. Genetics 163(3):1123–1134
Acknowledgments
This work was supported by China National Public Industrial (Agriculture) Specific Project (no. 201203091).
Author information
Authors and Affiliations
Corresponding author
Additional information
Daping Gong and Long Huang contributed equally to this article.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gong, D., Huang, L., Xu, X. et al. Construction of a high-density SNP genetic map in flue-cured tobacco based on SLAF-seq. Mol Breeding 36, 100 (2016). https://doi.org/10.1007/s11032-016-0514-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-016-0514-7