
Abstract
Field pea (Pisum sativum L.) is a grain legume crop that is cultivated for either human or livestock consumption. Development of varieties adapted to damaging abiotic and biotic stresses is a major objective for field pea breeding. Bacterial blight is a serious disease caused by the pathogenic agents Pseudomonas syringae pv. syringae and Pseudomonas syringae pv. pisi. A recombinant inbred line (RIL) genetic mapping population was generated by crossing the susceptible genotype Kaspa to the resistant genotype PBA Oura. Previously described sets of single-nucleotide polymorphism and simple sequence repeat markers were assigned to a genetic linkage map of the Kaspa × PBA Oura population, which contained 358 markers spanning 1070 cM with an average marker density of 1 locus per 3.0 cM. Combination with multiple previously published maps (including that of Kaspa × Parafield) subsequently generated an integrated structure with much higher marker density of 1 locus per 0.85 cM. The Kaspa × PBA Oura and Kaspa × Parafield RILs were screened at the seedling stage for resistance to both pathovars. Totals of four and two QTLs for resistance to infection by P. syringae pv. syringae were detected in the Kaspa × Parafield and Kaspa × PBA Oura populations, respectively. A single common genomic region associated with P. syringae pv. pisi resistance was identified in both mapping populations. To integrate information on bacterial blight resistance from various QTL mapping studies, the relevant regions were extrapolated on to the integrated map through use of common flanking markers. The resources generated in this study will support map enhancement, identification of marker-trait associations, genomics-assisted breeding, map-based gene isolation and comparative genetics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Field pea (Pisum sativum L.) is an annual cool-season legume crop with a global annual production of 11 million tonnes from 6.4 million hectares (FAOSTAT 2014). In many agricultural systems, legumes are complementary to cereals due to contributions from biological nitrogen fixation. Moreover, seeds of legumes contain valuable macro- and micro-nutrients, including proteins, providing a highly nutritious food resource (Duranti 2006). Production of field pea is constrained by the impact of a number of biotic and abiotic stresses, requiring improvement of levels of protection against major fungal, bacterial and viral diseases to increase yield and quality of the crop (Allen and Lenne 1998).
Bacterial blight, caused by the pathogenic agents P. syringae pv. syringae and P. syringae pv. pisi, is a sporadic and damaging disease of field pea. Incidence of bacterial blight has been reported in most of the pea-growing areas of the world. Prevalence of each pathogen varies between regions (depending on the predominant cultivar within that region), and they may occur in the field separately, or in combination (Martín-Sanz et al. 2011; Richardson and Hollaway 2011). A total of eight different pathogenic races of P. syringae pv. pisi have been described, based on their differential interaction with specific pea genotypes (Taylor et al. 1989; Martín-Sanz et al. 2011). In Australia, race 3 (64 %), followed by race 6 (31 %), is the most common of these races (Hollaway et al. 2007). P. syringae pv. pisi is the prevalent bacterial blight pathogen in Australian production zones, but the incidence of disease caused by P. syringae pv. syringae has increased since the introduction of semi-leafless cultivars (such as Kaspa, Excell and Snowpeak) in the south-eastern region (Richardson and Hollaway 2011).
Development of disease-resistant cultivars is the most effective method for control of bacterial blight, as chemical control is ineffective against both pathogens in the infected fields. Resistance to P. syringae pv. pisi is conferred by a specific gene-for-gene interaction mechanism, with six putative resistance–avirulence (R–A) gene pairs (Bevan et al. 1995). In contrast, resistance to P. syringae pv. syringae under controlled and field conditions exhibits continuous variation, suggesting contributions by a number of genes with lower magnitudes of effect, and leading to quantitative inheritance (Martín-Sanz et al. 2012). QTL studies for resistance to P. syringae pv. pisi in pea have identified several of these R genes (Ppi1 [R1] for race 1, Ppi2 [R2] for race 2, Ppi3 [R3] for race 3 and Ppi4 [R4] for race 4), which have been assigned to different genetic map locations (Ppi1 on linkage group [LG] VI, Ppi2 on LG VII, Ppi3 and Ppi4 probably on LG II [Hunter et al. 2001]). For P. syringae pv. pisi race 6, no source of resistance has so far been identified. In contrast, relatively few QTL studies have been conducted in order to identify mechanisms of resistance to bacterial blight caused by P. syringae pv. syringae, possibly due to limited availability of resistant genetic backgrounds (Fondevilla et al. 2012). A RIL population-based QTL analysis study identified quantitative variation for resistance, through detection of two QTLs accounting for 22 and 8 % of the phenotypic variation (Vp), respectively (Fondevilla et al. 2012).
Molecular genetic tools including high-throughput, robust molecular marker technologies such as SSR and SNP markers, high-density genetic linkage maps and consensus maps (Leonforte et al. 2013; Duarte et al. 2014; Sindhu et al. 2014; Sudheesh et al. 2014), and significant genomic resources such as transcriptome sequencing datasets (Franssen et al. 2011; Kaur et al. 2012; Duarte et al. 2014; Sudheesh et al. 2015) have been developed for pea, with an aim of crop improvement through enhancement of breeding programs. Genetic linkage maps are essential for QTL identification, marker-assisted selection, comparative genomics and map-based cloning (Semagn et al. 2006; McConnell et al. 2010), and also play a crucial role in anchoring physical maps and ordering and orientation of scaffolds during genome assemblies. Integration of multiple individual linkage maps into consensus structures permits saturation of marker content, with enhanced value across a broad range of germplasm sources, and ultimately allowing identification of the genetic factors that control agronomically important traits, including bacterial blight resistance, through QTL mapping and candidate gene identification.
The objectives of the present study were to develop a genetic linkage map for the Kaspa × PBA Oura RIL mapping population using SSR and SNP markers; to develop an integrated map of field pea by combining the Kaspa × PBA Oura map with previously described maps (Leonforte et al. 2013; Sudheesh et al. 2014; Duarte et al. 2014; Sindhu et al. 2014); and to detect QTLs associated with bacterial blight resistance in two RIL populations (Kaspa × Parafield and Kaspa × PBA Oura) that exhibit variation for the trait, using phenotypic data collected from glasshouse-based nursery screens.
Materials and methods
Plant materials
A segregating genetic mapping population was developed by crossing single genotypes from cultivar Kaspa (susceptible to both P. syringae pv. syringae and P. syringae pv. pisi race 3) to PBA Oura (moderately resistant to P. syringae pv. syringae and resistant to P. syringae pv. pisi race 3) (Richardson and Hollaway 2011) at the premises of Biosciences Research, Horsham, Victoria, Australia, and F2 progeny were obtained. Single-seed descent was performed for four subsequent generations in the glasshouse, and a total of 110 F6 RILs were subsequently generated. The plants were maintained in glasshouses under ambient conditions of 22 °C with a 16-/8-h (light/dark) photoperiod. Genomic DNA was extracted from leaves using the DNeasy® 96 Plant Kit (QIAGEN, Hilden, Germany) according to manufacturer’s instructions. The DNA was resuspended in MilliQ water and quantified using a Nanodrop spectrometer (Thermo Scientific, Massachusetts, USA), followed by storage at −20 °C.
SSR and SNP genotyping and genetic linkage mapping
SSR (Loridon et al. 2005; Kaur et al. 2012) and SNP (Leonforte et al. 2013) genotyping, followed by genetic linkage mapping and visualisation of the linkage groups of Kaspa × PBA Oura RILs, was performed as described previously (Leonforte et al. 2013; Sudheesh et al. 2014).
Integrated map construction
The Kaspa × PBA Oura genetic linkage map generated in this study was combined with the pre-existing Kaspa × Parafield (Leonforte et al. 2013), Kaspa × Yarrum and Kaspa × ps1771 (Sudheesh et al. 2014) maps to generate an intermediate structure, using MergeMap (Wu et al. 2011). Similarity searches of the DNA sequences underlying map-assigned SNP markers were performed against the transcriptome sequencing data (DDBJ/EMBL/GenBank under the accession GAMJ00000000 and KnowPulse—http://knowpulse.usask.ca/portal/search/sequences) underpinning the SNP markers on integrated maps of Duarte et al. (2014) and Sindhu et al. (2014) with a threshold E-value of 10−10. After identification of common sequences, data from all pre-existing maps were combined into a single integrated map structure through use of MergeMap. The linkage groups were visualised by use of MapChart (Voorrips 2002).
Phenotypic evaluation
Bacterial isolates
Bacterial isolates (P. syringae pv. syringae and P. syringae pv. pisi race 3) that were used in this study were obtained from Biosciences Research, Horsham, Victoria, Australia. The identities of the bacterial isolates were confirmed (using PCR), and bacterial isolates were maintained as described by Rodda et al. (2015).
Disease assessment
Screening for bacterial blight resistance in both mapping populations was performed under controlled environmental conditions at Biosciences Research, Horsham, Victoria, Australia. The experimental design was a randomised split block with three replicates of three plants each from each RIL-derived line in a pot. Two types of inoculation procedure (spray inoculation or stab inoculation) were performed on three- to four-week-old seedlings. P. syringae inoculum preparation and the inoculation procedures were performed as described previously [spray inoculation method—Rodda et al. (2015); stab inoculation method—Hollaway and Bretag (1995)]. Kaspa × Parafield RIL-derived plants were stab-inoculated with P. syringae pv. pisi race 3. Spray inoculation was performed for the other three combinations (Kaspa × PBA Oura and Kaspa × Parafield with P. syringae pv. syringae; Kaspa × PBA Oura with P. syringae pv. pisi race 3).
Following spray inoculation of P. syringae pv. syringae on both mapping populations, disease incidence scores were recorded as percentages of affected plant tissue. Following stab inoculation of the Kaspa × Parafield RIL population with P. syringae pv. pisi race 3, disease incidences were recorded on a numerical scale, with score 1—inoculation point dried, score 2—lesion growth arrested and score 3—water-soaked lesion. Means for disease scores were calculated, and if the value exceeded 2, a susceptible rating was assigned. In contrast, if the value was less than or equal to 2, a resistant rating was given. Following spray inoculation of the Kaspa × PBA Oura mapping population with P. syringae pv. pisi race 3, plants were scored on a scale of 1–4 (1—fully resistant; 2—moderately resistant; 3—moderately susceptible; and 4—fully susceptible). Mean scores for each individual from the mapping populations were used to construct frequency distribution histograms in order to determine mode of inheritance for each trait.
QTL analysis and identification of sequences associated with flanking genetic markers
The Kaspa × PBA Oura genetic linkage map that was developed in this study and Kaspa × Parafield genetic linkage map from Leonforte et al. (2013) were used for QTL analysis. QTL detection was performed as described by Sudheesh et al. (2014) using QTL Cartographer v 2.5 (Wang et al. 2012). QTLs identified in the current study and from a previous study (Fondevilla et al. 2012) were placed on the integrated map on the basis of common flanking markers.
BLASTN analysis of the sequences underlying SNP markers flanking the QTL-containing intervals was performed against the coding sequences (CDS) of M. truncatula, v3.5 (http://jcvi.org/medicago/), with a threshold E-value of 10−10. The sequences of SNP loci flanking the QTL-containing intervals were BLASTN analysed against the M. truncatula genome v3.5 (http://jcvi.org/medicago/), to identify genomic regions containing putative candidate genes.
Results
Genotyping and genetic linkage mapping
A total of 240 genomic DNA derived and EST-SSRs from different sources were screened to detect polymorphism between the parental lines of the Kaspa × PBA Oura mapping population, identifying 50 informative marker assays (40 genomic DNA derived and 10 EST-SSRs), of which 40 were successfully screened on 110 RILs. SNP genotyping was performed with 768 markers, of which 350 (46 %) revealed clear polymorphism and were hence processed for genetic linkage map construction. From the selected markers, a total of 23, including 9 SSRs and 14 SNPs, were excluded from linkage map construction due to skewed segregation ratios. The remaining 367 markers (31 SSRs and 336 SNPs) (Supplementary file 1) were used for linkage mapping, of which 358 (31 SSRs and 327 SNPs) were assigned to ten LGs (Supplementary files 2 and 3). The map spanned a total length of 1070 cM, with an average marker density of 1 locus per 3.0 cM (Supplementary file 4). The number of markers assigned to LGs varied from 5 (Ps VI) to 69 (Ps VII), with an average of 36 markers per LG. The identity and orientation of LGs were determined by comparison with the M. truncatula genome, as well as from previously map-assigned SSRs as anchoring markers. The largest numbers of SSR marker loci that were common between maps were on Ps IV and Ps VII, and the smallest was on Ps VI.
Comparative analysis of the four population-specific genetic maps (Kaspa × PBA Oura, Kaspa × Parafield, Kaspa × Yarrum and Kaspa × ps1771) revealed high levels of co-linearity, although specific map distances were not always comparable. A total of 123 markers were common across all four maps. The highest number of common markers (308) was between the Kaspa × Yarrum and Kaspa × ps1771 maps, followed by the comparison between the Kaspa × PBA Oura and Kaspa × ps1771 maps, with 281 markers. The lowest number of common markers was between the Kaspa × Yarrum and Kaspa × Parafield maps. Comparison of the common markers revealed co-linearity in locus order for Ps I, Ps III, Ps IV, Ps V and Ps VII. Alternatively, Ps II and Ps VI exhibited different genomic configurations, although co-linearity of gene order was maintained within the respective translocated segments. LGs containing large (≥20 cM) intervals devoid of any markers were observed for all four maps. The total number of gaps varied, the highest number (13) being for the Kaspa × Yarrum map, and the lowest (7) for the Kaspa × ps1771 and Kaspa × PBA Oura maps.
A composite structure was generated by merging information from the four maps, which were based on differing numbers of individual markers (358 in Kaspa × PBA Oura, 429 in Kaspa × Yarrum, 452 in Kaspa × ps1771 and 458 in Kaspa × Parafield). The composite map consisted of 782 markers (694 SNPs and 88 SSRs) which were assembled into seven LGs (Supplementary file 3), with a cumulative map length of 2601 cM. The lengths of LGs ranged from 293 cM (Ps VI) to 430 cM (Ps IV), with an average density of one marker per 3.4 cM. In some instances (37 in total, including three SSRs), the same marker assay detected multiple loci, which were consequently considered as unique markers. A total of 145 markers (19 %) were common to all four maps, while 234 (30 %) were unique to single populations (Kaspa × Parafield—128; Kaspa × Yarrum—63; Kaspa × ps1771—28 and Kaspa × PBA Oura—15), the remaining 548 (70 %) providing bridging loci between two or more maps. Comparison of the composite map with individual maps was performed to verify marker order in LGs, revealing high co-linearity in locus order (Supplementary file 5), apart from Ps II and Ps VI.
The DNA sequences underlying the 768 SNP loci were used for sequence similarity searches (Supplementary file 6) against pea transcriptome sequences underpinning those SNP markers assigned to previously published integrated maps. This process identified 203 bridging loci between the field pea composite map and the integrated maps. A comprehensive integrated structure was generated by merger of all available maps, permitting placement of 2857 markers on seven LGs spanning 2427 cM (Fig. 1; Table 1). Map density varied from 0.67 cM (Ps VI) to 1.09 cM (Ps II), with an average of 0.85 cM. Of the seven LGs, Ps VII contained the largest number of markers (525), followed by Ps III and Ps IV with 459, 392 markers, respectively, while Ps VI contained only 360 markers (Supplementary file 3). Some intervals devoid of any markers (>10 cM) were observed: single gaps on each of Ps I and Ps VII; three gaps on each of Ps II, Ps III and Ps IV, mainly towards the distal end; and a couple of gaps in the middle of each LG. Markers from each of the maps contributed unique loci to the integrated structure, and detailed analysis indicated that some of these loci were localised to specific regions. For instance, eight markers spanning 22 cM on the upper part of Ps V were derived from the composite structure of population-specific genetic maps. Marker order was well conserved in comparison with LGs of the individual maps, although several inversions and local rearrangements were observed.

Integrated map of field pea, with markers shown on the right of LGs, and distances between markers indicated in cM on the left. For presentation purposes, only one of a set of co-located genetic markers is shown on the map
Disease assessment and QTL detection
Significant differences between disease scores of whole plants following infection with P. syringae pv. syringae were observed between RILs of each population (Supplementary file 7). Frequency distribution patterns obtained from both populations indicated the presence of multiple genes contributing to disease resistance (Fig. 2). Evidence was also obtained for transgressive segregation patterns, as the phenotypic scores of some individual lines were located outside the range of the parental genotypes (being both more and less susceptible). The locations and magnitudes of effect for each QTL were estimated using both simple interval mapping (SIM) and composite interval mapping (CIM). On the Kaspa × PBA Oura map, two QTLs were detected (on Ps III and Ps VII), accounting for circa (c.) 15 % (Psy_KO1) and 11 % (Psy_KO2) of Vp, respectively (Fig. 3; Table 2). On the Kaspa × Parafield map, CIM detected four QTLs, two (Psy_KP2 and 3) of which were located on Ps III, accounting for c. 13 and 23 % of Vp, respectively (Table 2). The other two QTLs were detected on Ps II (Psy_KP4) and Ps VII (Psy_KP1), accounting for c. 9 and 8 % of Vp, respectively (Fig. 3, Table 2). For Psy_KP2, resistance determinants were derived from the susceptible parent (Kaspa), while the genetic factors at the other QTL regions were associated with the moderately resistant parent (Parafield).

Frequency distribution histograms generated from bacterial blight phenotyping using a percentage of infected area—P. syringae pv. syringae for Kaspa × Parafield; b percentage of infected area—P. syringae pv. syringae for Kaspa × PBA Oura; c disease scores—P. syringae pv. pisi race 3 for Kaspa × Parafield; and d disease scores—P. syringae pv. pisi race 3 for Kaspa × PBA Oura mapping populations

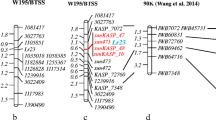
Localisation of QTLs associated with resistance to P. syringae pv. syringae and P. syringae pv. pisi race 3 on the a Kaspa × Parafield-derived genetic map; b Kaspa × PBA Oura-derived genetic map; and c integrated map. The name is provided at the top of each LG. Distances of the markers (cM) are shown to the left, and names of markers are shown to the right side of LGs. For presentation purposes, only selected markers are shown on the map
For the resistance to P. syringae pv. pisi race 3, only a single genomic region with moderate magnitude of effect was detected, located on Ps III of both the Kaspa × Parafield and Kaspa × PBA Oura maps, with maximum logarithm of odds (LOD) scores of 9.5 and 12.1, respectively, using CIM. The Ps III-located QTL accounted for c. 26 % (Kaspa × Parafield) and 38 % (Kaspa × PBA Oura) of Vp (Fig. 3, Table 2). The LOD peak for the QTL in both instances coincided with the marker SNP_100000801, further supporting the inference that a single common genomic region is responsible for conferring resistance.
All QTLs identified in the current study, as well those from a previous study (Fondevilla et al. 2012), were extrapolated onto the integrated map. This process obtained seven loci distributed across three LGs, with number of loci per LG varying from 1 to 4 (Fig. 3). A single correspondence was observed with the previous study, based on the location of the PsBB2_Psy from the Kaspa × Parafield population.
Annotation of the flanking markers
Comparison of DNA sequences associated with markers linked to the P. syringae pv. syringae resistance QTLs directly identified M. truncatula CDS with functional annotations (supplementary file 8) such as serine/threonine-protein kinase, monothiol glutaredoxin-S15, polyadenylate-binding protein 2, cysteine proteinase 3 and serine hydroxymethyltransferase gene products, some of which have been reported to be involved in disease resistance mechanisms of plants. Similarly, the sequences associated with SNP markers flanking the P. syringae pv. pisi race 3 resistance QTL-containing intervals included annotations (supplementary file 8) as ascorbate peroxidase, rhodanese-like and caffeic acid O-methyltransferase gene products.
Moreover, regions of the M. truncatula genome near and within the QTL-containing intervals were examined for candidate gene presence. Multiple genes located in the vicinity of QTLs (Psy_KP3, Psy_KO1, Psy_KO 2 and Ppi3) on Ps III (Mt III) and Ps VII (Mt IV and Mt VIII) were annotated as being involved in disease resistance (Supplementary file 9).
Discussion
Genotyping and linkage mapping
In the recent past, several field pea genetic linkage maps have been developed, mostly through the use of SNP and SSR marker technologies (Leonforte et al. 2013; Duarte et al. 2014; Sindhu et al. 2014; Sudheesh et al. 2014). The Kaspa × PBA Oura linkage map constructed in the present study exhibits an average marker density of 1 per 3.0 cM and a cumulative length of 1070 cM. This value is comparatively smaller than those of the maps from Leonforte et al. (2013) and Sudheesh et al. (2014), possibly due to the effects of a lower number of map-assigned markers in the present study (358). Comparisons with previously constructed maps (Leonforte et al. 2013; Sudheesh et al. 2014) revealed highly conserved marker orders, although the corresponding distances were not always in similar proportion.
Individual genetic linkage maps exhibit limitations such as genotype-specific chromosomal rearrangements (duplications, translocations, inversions), restricted access to population-specific information and limitations of marker content and density (Yu et al. 2010). Combination of data from multiple linkage maps into a single integrated structure can address these shortcomings and facilitate the comparison of QTL regions identified in various genetic backgrounds. In the present study, this objective was accomplished in a two-step process, initially by merger of four population-specific genetic linkage maps and then by combination with two other integrated maps. An integrated map has a higher level of relevance for positioning of the order of markers than to determine absolute distances between markers. The recombination frequencies of maps from different mapping populations are not fully consistent, although the MergeMap product provided a more accurate description of marker order than a linear map derived using approximations based on recombination values (Close et al. 2009). Moreover, the consensus map based on merging different maps is simply one of many possible non-conflicting linear representations of the consensus DAGs. MergeMap software, which was used for this purpose, has been used in construction of consensus maps in crops such as common bean (Phaseolus vulgaris L.) (Galeano et al. 2012), peanut (Arachis hypogaea L.) (Gautami et al. 2012) and faba bean (Vicia faba L.) (Satovic et al. 2013). The integrated map contained 829 more markers than a previously constructed consensus map (Sudheesh et al. 2014). Although high levels of co-linearity were observed between the two maps, addition of new markers generated a small number of fine-scale differences.
Disease assessment, QTL detection and annotation of the flanking markers
The present study used two different methodologies for phenotypic assessment of disease resistance. Previously, the stab inoculation method has been used for bacterial inoculation (Hollaway and Bretag 1995), but is a slow and laborious process and not suitable for use with P. syringae pv. syringae. The spray inoculation method more closely reflects the natural means of infection than stab inoculation, and is also more efficient and reproducible (Rodda et al. 2015). Only minor differences in disease symptoms due to different inoculation methods were observed.
In the present study, phenotypic screening for bacterial blight resistance was conducted under controlled conditions in the glasshouse using a single P. syringae pv. syringae isolate. A significant challenge arises from the presence of partial resistance effects which require quantitative assessment, in contrast to major R genes that generally give rise to qualitative effects on the phenotype. Only limited studies to date have been conducted to analyse the resistance of pea to P. syringae pv. syringae, under both controlled and field conditions (Martín-Sanz et al. 2011; Richardson and Hollaway 2011). A continuous frequency distribution of resistance scores in response to P. syringae pv. syringae infection was observed for both RIL populations in the present study, suggesting quantitative inheritance. An earlier study in pea also reported a continuous distribution, supporting this hypothesis (Fondevilla et al. 2012).
CIM analysis detected totals of 4 and 2 QTLs for resistance to bacterial blight caused by P. syringae pv. syringae in the Kaspa × Parafield and Kaspa × PBA Oura mapping populations, respectively. This discrepancy may be due to the differences of genetic background. For the Kaspa × Parafield population, both parents contributed alleles for resistance. The inheritance of such favourable alleles from susceptible parents has been reported previously (Tar’an et al. 2004). For the Kaspa × PBA Oura RIL population, only the resistant parent (PBA Oura) contributed alleles for bacterial blight resistance. It is possible that such differences may have arisen from gene interaction effects, especially due to a predominant effect of genetic contributions of a more resistant parent (PBA Oura).
The parental lines of the two mapping populations exhibited relatively narrow differences in resistance level, and this may have contributed to the identification of QTLs accounting for low-to-moderate proportions of Vp, rather than major gene effects, which would generally be manifested through highly divergent performance of parental genotypes. In both mapping populations, transgressive lines with higher resistance than the parents were observed. This observation indicates the potential to select such lines, which may provide a novel source of resistance in breeding programs.
QTL analysis for P. syringae pv. pisi resistance revealed a single locus of moderate effect on the genetic map of the Kaspa × Parafield mapping population. A similar result was obtained for the Kaspa × PBA Oura population. The detailed genetic map structure and marker content vary between the two populations, but the presence of a common flanking marker strongly suggests that the same genomic region is involved. Identification of the same genomic region in both mapping populations supports the view that differences between inoculation methodologies did not contribute to variability of the disease development in the RILs. Differing proportions of Vp accounted for the Ppi3 QTLs detected from the two mapping populations may be due to a number of contributory factors, including differences in genetic background between the two populations; difference in levels of resistance directly conferred by the causal gene(s) that contribute the QTLs, possibly due to independent allelic effects; and methodological differences of phenotypic screening and assessments. In support of these possibilities, there is no commonality of genetic background between the two resistant parents, Parafield and PBA Oura.
Previous studies to evaluate responses to P. syringae pv. pisi revealed that race-specific resistance under controlled conditions for races 2, 3 and 4 was also observed under field conditions (Martín-Sanz et al. 2012). This outcome suggests that the corresponding resistance genes (R2, R3 and R4) are effective under genuine field conditions. Moreover, the gene combination (R2 + R3 + R4) provided a more effective resistance than a single gene in controlling most P. syringae pv. pisi races, including the newly identified race 8, but was ineffective against race 6 (Elvira-Recuenco et al. 2003; Martín-Sanz et al. 2012). The major class of R genes are dominant in nature (De Ronde et al. 2014), and so highly suitable for use in marker-assisted selection (Tiwari and Singh 2012) and gene pyramiding. Previous studies (Martín-Sanz et al. 2012; Rodda et al. 2015) showed that the outcomes of phenotyping screening conducted under controlled conditions were positively correlated with field-based disease symptoms, and glasshouse-based leaf inoculation provides a useful method for the prediction of response under field conditions. For example, consistent resistance ratings were observed for the field pea cultivar PBA Percy under both glasshouse and field conditions while infected with P. syringae pv. syringae (Rodda et al. 2015).
For comparative purposes, the bacterial blight resistance QTLs were placed on the integrated map through use of common flanking markers. The incorporation of information from multiple independent QTL mapping studies provided an enhanced assessment of the genetic basis for resistance. Previous literature had reported the map positions of two QTLs determining resistance to P. syringae pv. syringae (Fondevilla et al. 2012). However, only one of these QTLs shared linked markers in common with the present study, revealing co-localisation with PsBB2_Psy on the integrated map. Identification of common QTLs between different segregating populations provides increased confidence in prospects for marker-assisted breeding, as well as allowing determination of a set of candidate markers for further enrichment of bacterial blight resistance QTL-containing regions on population-specific genetic maps in future. The high levels of concordance observed between marker locations in the integrated map and those of the population-specific genetic linkage maps confirm the value of the approach.
Functional annotation of sequences underpinning the genetic markers that flank the relevant QTL-containing intervals successfully identified genes associated with plant defence mechanisms in field pea. Serine/threonine kinases (SNP_100000290, Psy_KP1 region) play a role in recognition of pathogen-derived signal molecules and also different signalling levels in the context of non-race-specific elicitation, gene-for-gene interactions, or resistance to virulent pathogens in a number of different systems including tobacco, tomato, alfalfa, parsley, rice and Arabidopsis thaliana (Romeis 2001). Plant cysteine proteases (SNP_100000076, Psy_KP4 region) are proteolytic enzymes which are closely regulated during physiological functions (growth, development and accumulation–mobilization of storage proteins) and defence roles (Martínez et al. 2012).
Similarity searches of sequences associated with P. syringae pv. pisi race 3 QTL region-flanking markers against M. truncatula CDS facilitated identification of candidate genes such as plant ascorbate peroxidase (SNP_100000801, SNP_100000802 and SNP_100000347) and caffeic acid O-methyltransferase (SNP_100000063). Elevated expression of enzymes such as ascorbate peroxidase is also observed during defence mechanism of plants against a broad range of pathogens. Caffeic acid O-methyltransferase performs a key role in the biochemical pathway leading to lignin subunit (monolignol) synthesis (Ma and Xu 2008; Caverzan et al. 2012). During pathogen attack, defence reactions such as lignin production are activated, as a well-characterised mechanism of plant cell wall reinforcement against pathogen entry (Shimada et al. 2000).
The comparative analysis of the sequences underpinning the SNP markers flanking the QTL intervals (on Ps III and Ps VII) with the M. truncatula genome revealed the presence of multiple disease resistance genes within those intervals. A high level of conserved synteny was observed between the pea genetic map and the M. truncatula genome (Leonforte et al. 2013). It is therefore possible that the identified QTL regions may be conserved between these species and that the genes identified within the QTL-containing regions may be plausible candidates, although additional studies will be required for validation.
Implications for field pea breeding programs
The present study has developed valuable resources for pea linkage mapping, especially the integrated map which will enhance future mapping analysis studies and marker implementation in pea breeding programs. Important sources of bacterial blight resistance in field pea were identified, and the genetic markers flanking the QTL-containing regions identified in this study can be used for the development of linked and diagnostic polymorphisms for marker-assisted selection (MAS) of resistant cultivars, based on introgression of QTL-containing genomic regions from donor to recipient germplasm. Moreover, as QTLs for resistance to both P. syringae pv. syringae and P. syringae pv. pisi race 3 are co-located on Ps III of Kaspa × PBA Oura, this region is an important target for improvement of bacterial blight resistance and provides the basis for co-selection in genomics-assisted breeding practice. Pea breeding programs that aim to develop bacterial blight resistance cultivars should target a combination of race-specific and non-specific resistance, in order to provide an optimised genetic background for protection against the disease.
References
Allen DJ, Lenne JM (1998) The pathology of food and pasture legumes. CAB International, Wallingford. ISBN 0-85199-166-1
Bevan JR, Taylor JD, Crute IR, Hunter PJ, Vivian A (1995) Genetics of specific resistance in pea (Pisum sativum) cultivars to seven races of Pseudomonas syringae pv. pisi. Plant Pathol 44:98–108
Caverzan A, Passaia G, Rosa SB, Ribeiro CW, Lazzarotto F, Margis-Pinheiro M (2012) Plant responses to stresses: role of ascorbate peroxidase in the antioxidant protection. Genet Mol Biol 35:1011–1019
Close TJ, Bhat PR, Lonardi S, Wu Y, Rostoks N et al (2009) Development and implementation of high-throughput SNP genotyping in barley. BMC Genom 10:582. doi:10.1186/1471-2164-10-582
De Ronde D, Butterbach P, Kormelink R (2014) Dominant resistance against plant viruses. Front Plant Sci 5:307. doi:10.3389/fpls.2014.00307
Duarte J, Rivière N, Baranger A, Aubert G, Burstin J, Cornet L, Lavaud C, Lejeune-Hénaut I, Martinant JP, Pichon JP, Pilet-Nayel ML, Boutet G (2014) Transcriptome sequencing for high throughput SNP development and genetic mapping in pea. BMC Genom 15:126
Duranti M (2006) Grain legume proteins and nutraceutical properties. Fitoterapia 77:67–82
Elvira-Recuenco M, Bevan JR, Taylor JD (2003) Differential responses to pea bacterial blight in stems, leaves and pods under glasshouse and field conditions. Eur J Plant Pathol 109:555–564
FAOSTAT (2014). http://faostat3.fao.org/home/E. Accessed 3 April 2015
Fondevilla S, Martín-Sanz A, Satovic Z, Fernández-Romero M, Rubiales D, Caminero C (2012) Identification of quantitative trait loci involved in resistance to Pseudomonas syringae pv. syringae in pea (Pisum sativum L.). Euphytica 186:805–812
Franssen SU, Shrestha RP, Bräutigam A, Bornberg-Bauer E, Weber APM (2011) Comprehensive transcriptome analysis of the highly complex Pisum sativum genome using next generation sequencing. BMC Genom 12:227
Galeano CH, Cortés AJ, Fernandez AC, Soler A, Franco-Herrera N et al (2012) Gene-based single nucleotide polymorphism markers for genetic and association mapping in common bean. BMC Genet 13:48
Gautami B, Foncéka D, Pandey MK, Moretzsohn MC, Sujay V et al (2012) An international reference consensus genetic map with 897 marker loci based on 11 mapping populations for tetraploid groundnut (Arachis hypogaea L.). PLoS ONE 7:e41213
Hollaway GJ, Bretag TW (1995) Occurrence and distribution of races of P. syringae pv. pisi in Australia and their specificity towards various field pea (Pisum sativum) cultivars. Aust J Exp Agr 35:629–632
Hollaway GJ, Bretag TW, Price TV (2007) The epidemiology and management of bacterial blight (Pseudomonas syringae pv. pisi) of field pea (Pisum sativum) in Australia: a review. Aust J Agr Res 58:1086–1099
Hunter PJ, Ellis N, Taylor JD (2001) Association of dominant loci for resistance to Pseudomonas syringae pv. pisi with linkage groups II, VI and VII of Pisum sativum. Theor Appl Genet 103:129–135
Kaur S, Pembleton L, Cogan NOI, Savin KW, Leonforte T, Paull J, Materne M, Forster JW (2012) Transcriptome sequencing of field pea and faba bean for discovery and validation of SSR genetic markers. BMC Genom 13:104
KnowPulse (2015). http://knowpulse.usask.ca/portal/search/sequences. Accessed 3 April 2015
Leonforte A, Sudheesh S, Cogan NOI, Salisbury PA, Nicolas ME, Materne M, Forster JW, Kaur S (2013) SNP marker discovery, linkage map construction and identification of QTLs for enhanced salinity tolerance in field pea (Pisum sativum L.). BMC Plant Biol 13:161
Loridon K, McPhee K, Morin J, Dubreuil P, Pilet-Nayel ML, Aubert G, Rameau C, Baranger A, Coyne C, Lejeune-Hènaut I, Burstin J (2005) Microsatellite marker polymorphism and mapping in pea (Pisum sativum L.). Theor Appl Genet 111:1022–1031
Ma QH, Xu Y (2008) Characterization of a caffeic acid 3-O-methyltransferase from wheat and its function in lignin biosynthesis. Biochimie 90:515–524
Martínez M, Cambra I, González-Melendi P, Santamaría ME, Díaz I (2012) C1A cysteine-proteases and their inhibitors in plants. Physiol Plant 145:85–94. doi:10.1111/j.1399-3054.2012.01569.x
Martín-Sanz A, Palomo JL, Pérez de la Vega M, Caminero C (2011) Identification of pathovars and races of Pseudomonas syringae, the main causal agent of bacterial diseases in pea in North-Central Spain, and the search for resistance. Eur J Plant Pathol 129:57–69
Martín-Sanz A, Pérez de la Vega M, Caminero C (2012) Resistance to Pseudomonas syringae in a collection of pea germplasm under field and controlled conditions. Plant Pathol 61:375–387
McConnell M, Mamidi S, Lee R, Chikara S, Rossi M, Papa R, McClean P (2010) Syntenic relationships among legumes revealed using a gene-based genetic linkage map of common bean (Phaseolus vulgaris L.). Theor Appl Genet 121:1103–1116. doi:10.1007/s00122-010-1375-9
Medicago Truncatula Genome Project. http://jcvi.org/medicago/. Accessed 3 April 2015
Richardson HJ, Hollaway GJ (2011) Bacterial blight caused by Pseudomonas syringae pv. syringae shown to be an important disease of field pea in south eastern Australia. Australas Plant Path 40:260–268
Rodda MS, Kant P, Lindbeck KD, Gnanasambandam A, Hollaway GJ (2015) A high-throughput glasshouse based screening method to evaluate bacterial blight resistance in field pea (Pisum sativum). Aust Plant Pathol. doi:10.1007/s13313-015-0374-3
Romeis T (2001) Protein kinases in the plant defence response. Curr Opin Plant Biol 4:407–414
Satovic Z, Avila CM, Cruz-Izquierdo S, Díaz-Ruíz R, García-Ruíz GM, Palomino C, Gutiérrez N, Vitale S, Ocaña-Moral S, Gutiérrez MV, Cubero JI, Torres AM (2013) A reference consensus genetic map for molecular markers and economically important traits in faba bean (Vicia faba L.). BMC Genom 14:932. doi:10.1186/1471-2164-14-932
Semagn K, Bjørnstad A, Ndjiondjop MN (2006) Principles, requirements and prospects of genetic mapping in plants. Afr J Biotechnol 5:2569–2587
Shimada N, Akashi T, Aoki T, Ayabe S (2000) Induction of isoflavonoid pathway in the model legume Lotus japonicus: molecular characterization of enzymes involved in phytoalexin biosynthesis. Plant Sci 160:37–47
Sindhu A, Ramsay L, Sanderson L-A, Stonehouse R, Li R, Condie J, Shunmugam ASK, Liu Y, Jha AB, Diapari M, Burstin J, Aubert G, Tar’an B, Bett KE, Warkentin TD, Sharpe AG (2014) Gene-based SNP discovery and genetic mapping in pea. Theor Appl Genet 127:2225–2241
Sudheesh S, Lombardi M, Leonforte A, Cogan NOI, Materne M, Forster JW, Kaur S (2014) Consensus genetic map construction for field pea (Pisum sativum L.), trait dissection of biotic and abiotic stress tolerance and development of a diagnostic marker for the er1 powdery mildew resistance gene. Plant Mol Biol Rep. doi:10.1007/s11105-014-0837-7
Sudheesh S, Sawbridge TI, Cogan NOI, Kennedy P, Forster JW, Kaur S (2015) De novo assembly and characterisation of the field pea transcriptome using RNA-Seq. BMC Genom 16:611. doi:10.1186/s12864-015-1815-7
Tar’an B, Warkentin T, Somers DJ, Miranda D, Vandenberg A, Blade S, Woods S, Bing D (2004) Identification of quantitative trait loci for grain yield, seed protein concentration and maturity in field pea (Pisum sativum L). Euphytica 136:297–306
Taylor JD, Bevan JR, Crute IR, Reader SL (1989) Genetic relationship between races of Pseudomonas syringae pv. pisi and cultivars of Pisum sativum. Plant Pathol 38:364–375
Tiwari JK, Singh BP (2012) Marker-assisted selection for virus resistance in potato: options and challenges. Potato J 39:101–117
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78
Wang S, Basten CJ, Zeng ZB (2012) Windows QTL Cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh, NC. http://statgen.ncsu.edu/qtlcart/WQTLCart.htm
Wu Y, Close TJ, Lonardi S (2011) Accurate construction of consensus genetic maps via integer linear programming. IEEE/ACM Trans Comput Biol Bioinform 8:381–394. doi:10.1109/TCBB.2010.35
Yu J, Kohel RJ, Smith CW (2010) The construction of a tetraploid cotton genome wide comprehensive reference map. Genomics 95:230–240
Acknowledgments
This work was supported by funding from the Victorian Department of Economic Development, Jobs, Transport and Resources and the Grains Research and Development Council, Australia.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
11032_2015_376_MOESM1_ESM.xlsx
Supplementary material 1 Marker information: This file contains information on the total number of markers analysed, tested for polymorphism, used for linkage mapping and assigned to genetic linkage map locations. (XLSX 9 kb)
11032_2015_376_MOESM2_ESM.pptx
Supplementary material 2 Kaspa × PBA Oura linkage map of field pea: This file contains the visual representation of linkage maps of field pea based on genotyping of Kaspa × PBA Oura RILs. Names are provided at the top of each LG. Distances of the markers (cM) are shown to the left, and names of markers are shown to the right side of the linkage groups. For presentation purposes, only one of a set of co-located genetic markers are shown on the map. (PPTX 190 kb)
11032_2015_376_MOESM3_ESM.xlsx
Supplementary material 3 Linkage map statistics: This file contains details of different markers (SSRs and SNPs) and their corresponding positions on different LGs of (Tab 1) the Kaspa × PBA Oura RIL mapping population; (Tab 2) Composite map; (Tab 3) Integrated map. (XLSX 151 kb)
11032_2015_376_MOESM4_ESM.xlsx
Supplementary material 4 Marker distribution over the LGs of the Kaspa × PBA Oura map: This file contains the information about the distribution of the markers over the LGs of Kaspa × PBA Oura map. (XLSX 9 kb)
11032_2015_376_MOESM5_ESM.pptx
Supplementary material 5 Comparison between linkage group (Ps V) of composite map with Kaspa × PBA Oura, Kaspa × Parafield, Kaspa × Yarrum and Kaspa × ps1771: This file shows the visual representation of the comparison between Ps V from the composite map with the contributing Kaspa × PBA Oura, Kaspa × Parafield, Kaspa × ps1771 and Kaspa × Yarrum maps, and the common markers between them. Pink coloured lines represent the corresponding positions of common markers. (PPTX 217 kb)
11032_2015_376_MOESM6_ESM.xlsx
Supplementary material 6 BLAST results: The Excel file contains the BLAST-based sequence analysis of (Tab 1) DNA sequences underlying SNP markers from Leonforte et al. (2013) against the transcriptome sequencing data from Sindhu et al. (2014); (Tab 2) DNA sequences underlying SNP markers from Leonforte et al. (2013) against the transcriptome sequencing data from Duarte et al. (2014); (Tab 3) Transcriptome sequencing data from Duarte et al. (2014) against the Sindhu et al. (2014). (XLSX 3780 kb)
11032_2015_376_MOESM7_ESM.pptx
Supplementary material 7 Bacterial blight disease figures: Disease symptoms observed on Kaspa × Parafield RIL-derived plants following inoculation with P. syringae pv. syringae: (a) 50 % of the plant infected; (b) 60 % of the plant infected; (c) 90 % of the plant infected; and (d) 100 % of the plant infected. (PPTX 480 kb)
11032_2015_376_MOESM8_ESM.xlsx
Supplementary material 8 Annotation of QTL flanking markers: This file contains annotations for the sequences underlying the SNP markers flanking the QTL intervals. (XLSX 10 kb)
11032_2015_376_MOESM9_ESM.xlsx
Supplementary material 9 Comparative analysis of QTL intervals with M. truncatula genome: This file contains comparative analysis of the QTL intervals of field pea genetic maps with M. truncatula genome. (XLSX 168 kb)
Rights and permissions
About this article

Cite this article
Sudheesh, S., Rodda, M., Kennedy, P. et al. Construction of an integrated linkage map and trait dissection for bacterial blight resistance in field pea (Pisum sativum L.). Mol Breeding 35, 185 (2015). https://doi.org/10.1007/s11032-015-0376-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-015-0376-4