
Abstract
Flavour is an important food trait, yet little is known about the genetic architecture and mode of inheritance of apple flavour compounds. The objectives of this study were to: understand the inheritance of flavour volatiles in a clonally replicated germplasm population; unravel correlation networks of volatiles; and to use genome-wide single nucleotide polymorphism (SNP) markers to identify genomic regions that play a role in the expression of flavour volatiles. This analysis revealed that more than half of the 37 volatiles (measured by gas chromatography–mass spectrometry) showed high heritability (h 2 > 0.4), with only a small number (3 of the 37) displaying low heritability (h 2 < 0.2). Majority (~85 %) of the significant SNP loci displayed the additive mode of inheritance. Our results supported the roles of MdAAT, MdCXE and MdLOX genes in the expression of apple flavour volatiles. Effect sizes of SNP loci, some of which are associated with multiple compounds, were small (<10 %), which is consistent with a polygenic quantitative inheritance model. New genomic locations associated with multiple flavour compounds were found, and some SNPs were associated with both sensory flavour and some flavour volatiles. Simultaneous genome-wide association study for sensory flavour and flavour compounds is recommended for unravelling genetic mechanisms to facilitate marker-assisted breeding for targeted flavours.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The most important objective across apple breeding programmes has been to increase the marketability of the fruit by improving their appearance, eating quality and flavour (Laurens 1999). Flavour is a complex trait resulting from combinations of sugars, acids and many volatile chemicals (Dixon and Hewett 2000). Over 300 volatile compounds have been measured in the aroma profile of apples (Dimick and Hoskin 1983; Paillard 1990). For ease of measurement, it is a common practice in apple breeding programmes to assess overall ‘sensory flavour intensity’ (FINT) using ordinal scores [e.g. 0 (none) to 9 (highest)] for segregating seedlings (Durel et al. 1998). Although a great range of volatile compounds may contribute to the flavour type and intensity of fresh apple flavour, the majority are esters (e.g. ethyl butanoate, ethyl 2-methylbutanoate, butyl acetate, 2-methylbutyl acetate and hexyl acetate) and alcohols (e.g. butanol, hexanol and trans-2-hexanol) (Paillard 1990; Dunemann et al. 2012).
Some volatiles also correlate with sweet taste, sour taste, as well as with consumer acceptability (Baldwin et al. 1998). Karlsen et al. (1999) reported that the main volatile components contributing to the sensory flavour profiles of some common apple cultivars were propyl acetate, butyl acetate, 2-methylbutyl acetate, hexyl acetate, ethyl acetate, ethyl butanoate, 1-butanol, ethanol and α-farnesene. The first four of these volatiles were related to fruity and sweet sensory attributes, while the rest were related to sour, bitter and grassy attributes. Mehinagic et al. (2006) also reported association of 2-methylbutyl acetate and hexyl acetate with sensory descriptors such as fruity and sweet. Fruit soluble solids concentration (SSC) is the best objective predictor of sensory sweetness (Harker et al. 2002), and one strategy for breeding new apple cultivars with high FINT is to increase soluble solids contents.
There is growing interest in understanding the genetic basis of specific flavour volatiles in order to implement marker-assisted selection (MAS) for flavour. Quantitative trait locus (QTL) mapping studies for flavour volatiles either used bi-parental families (Zini et al. 2005; Dunemann et al. 2009; Rowan et al. 2009a; Costa et al. 2013; Souleyre et al. 2014) or screened apple cultivars using a few candidate genes (Dunemann et al. 2012; Ulrich and Dunemann 2012; Vogt et al. 2013). QTLs identified in bi-parental families generally lack repeatability across genetic backgrounds, and the effect sizes are generally overestimated largely because of small sample sizes (Hayes and Goddard 2001; Flint and Mackay 2009).
Functional genomics studies have also been conducted to understand the roles that different genes play in the biosynthesis of flavour volatiles, especially esters. A number of alcohol acyl transferase (AAT) genes have been identified and characterised for their putative biochemical roles and expression patterns (Souleyre et al. 2005; Zhu et al. 2008; Dunemann et al. 2012). MpAAT1 encodes an enzyme capable of catalysing the synthesis of the main esters in ripe ‘Royal Gala’ fruit from a broad range of alcohol and coenzyme A (CoA) substrates (Souleyre et al. 2005). Expression of the highly homologous gene MdAAT2 is also positively correlated with AAT enzyme activity and ester production in ‘Golden Delicious’ (Li et al. 2006). Souleyre et al. (2014) provided evidence that structural differences between ‘Royal Gala’ and ‘Granny Smith’ AAT1 enzymes could account for the qualitative and quantitative differences in ester profiles. Stable transgenic AAT1 down-regulated ‘Royal Gala’ plant also indicated that AAT1 had a strong influence on the levels of esters and the pool size of ester precursors in ‘Royal Gala’ fruit (Souleyre et al. 2014). Nieuwenhuizen et al. (2013) showed that the cultivated apple genome contains only 10 terpene synthase (TPS) functional genes, and eight of these genes could account for the majority of terpene volatiles production in ‘Royal Gala’. Carboxylesterases (CXE) enzymes catalyse the hydrolysis of esters, and MdCXE1 gene has been suggested to play an important role in ester metabolism of apple fruit (Souleyre et al. 2011). Despite various QTL mapping and functional genomics studies, there are no applications of marker-assisted selection (MAS) for flavour in apple breeding programmes.
There is a lack of basic understanding of the inheritance, the interplay of genetics and environment and the genetic relationships among different apple flavour volatiles. Such knowledge is essential, along with the identification of genomic regions, which contribute to the observed phenotypic variability of flavour volatiles to breed new cultivars with particular flavour. Understanding of genetic architecture is necessary to choose the best strategy (MAS or genomic selection) for genetic improvement of a trait and to provide some lead to conduct studies for functional validation of genomic hot spots. The objectives of this study include: the understanding of inheritance patterns of flavour volatiles measured by gas chromatography–mass spectrometry (GC–MS) in a clonally replicated population of wide genetic background; identifying correlation networks of volatiles; and the use of a single nucleotide polymorphism (SNP) array to identify genomic regions that are important in the biosynthesis of flavour volatiles. The 8K SNP array used in this study encompassed some candidate genes, so we also validated the effect size of these genes in a wide genetic background.
Materials and methods
Plant material
Broadening of the Malus gene pool in New Zealand was initiated by establishing a highly diverse apple breeding population. The first generation of this population was established by planting nearly 30,000 open-pollinated (OP) seedlings of nearly 500 primitive apple cultivars and some wild species from around the world during the early 1990s (Kumar et al. 2010). About 300 seedlings, identified using data on tree, fruit and disease resistance traits, were then mated to generate nearly 9000 second-generation seedlings. Following the collection of fruit traits data, a diverse set of 230 seedlings was identified (Kumar et al. 2010). Two copies (or clones) of each of these 230 individuals were then clonally propagated on to ‘M.9’ rootstock and planted in a common-garden experiment in 2009 on a Plant & Food Research orchard (39°40′S 176°53′E) at the Hawke’s Bay Research Centre, New Zealand. The ‘status number’ (which is also interpreted as the effective population size; see Lindgren and Mullin 1998) of these 230 individuals was 72—suggesting considerable relatedness. The average pair-wise coefficient of relationship (= twice the coefficient of co-ancestry) among these individuals was 0.03, while 10 and 1 % of all pairs were half-sibs and full-sibs, respectively. Currently, these individuals are being characterised for a range of traits including the flavour volatiles reported in this study.
Phenotypes
Flavour volatile measurements were obtained on fruit from 200 individuals (or genotypes) over a period of three years. Of the 200 individuals, 58, 76 and 46 were measured only once in 2011, 2012 and 2013, respectively. There were 20 individuals assessed in each of the three seasons/years (2011–2013) to estimate seasonal effect and adjust phenotypes of other individuals for such effects. Each individual was represented by two clonal replicates, so altogether there were 480 measurements for each volatile. Titratable acidity (TA) and SSC were also obtained on fruit from each seedling using an automatic acid titrator (Metrohm 716 DMS) and a digital refractometer (Atago PR-32), respectively.
Fruit harvesting, which was carried out in the fruiting season (February–May), began when fruit from each tree was judged to be mature, based on a change in skin background colour from green to yellow, and when the starch pattern index was between 2 and 3 using a scale from 0 (full starch) to 7 (no starch). Separate samples of six fruit from two clonal replicates of each genotype were stored for 42 days at 0.5 °C and then a further 1 day at 20 °C before evaluation. The volatile analysis procedure using GC–MS, as described by Rowan et al. (2009b), was used except that fruit was placed in 4-L unused commercial metal paint cans rather than glass jars. After volatile collection, the absorbent traps were eluted with diethyl ether (2 × 1 mL) containing tetradecane at 10 nL mL−1 into pre-weighed 4-mL glass vials at a flow rate of 2 mL min−1. Samples were stored at −20 °C before GC–MS analysis. Volatiles were identified from their retention index and by comparison with commercial mass spectral databases and authentic compounds. Generally, base peak intensities were used to aid automated peak identification and integration using Waters QuanLynx software. Fruit volume, and hence surface area, was calculated, and volatile concentrations are reported as ng tetradecane (m/z 57) equivalents released cm−2 fruit surface area per hour.
Estimation of genetic parameters
All volatile concentrations were log10 transformed before further analysis to improve normality. Observations on clonal replicates of an individual were treated as repeated measurements, so it was desirable to model ‘permanent environment effects’ to prevent upward bias in additive genetic variance. We used the following mixed linear model accounting for repeated records for each trait:
where y is the vector of observations, b is the vector of fixed effects (e.g. fruit surface area, year/season of assessment), a is a vector of additive genetic effects of an individual, p is a vector of permanent environmental effects and e is a vector of residual effects. The matrix X is the incidence matrix for the fixed effects and Z is the incidence matrix relating observations to seedlings. Each individual has an additive genetic as well as a permanent environmental effect, so both effects have the same design matrix (Z). The associated variances with the random effects, a, p and e, were \(\sigma_a^2\), \(\sigma_p^2\) and \(\sigma_e^2\), respectively. We assumed that within an individual there was no correlation between its additive and its permanent environmental effect and that permanent environmental effects for different seedlings were uncorrelated. Estimates of narrow-sense heritability (h 2) of each trait were obtained as the ratio of additive variance (\(\sigma_a^2\)) to the total phenotypic variance (= \(\sigma_a^2 + \sigma_p^2 + \sigma_e^2\)). Genetic relationships among individuals were taken into account via the additive genetic relationship matrix. Also obtained from Eq. 1 were best linear unbiased prediction (BLUP) of additive genetic values (i.e. a) of all individuals for all volatiles, and then, between-volatile additive genetic correlations were estimated as product–moment correlation between BLUPs. To calculate phenotypic correlations between volatiles, we used best linear unbiased estimates (BLUEs) obtained by treating individuals effect (a) as fixed. Equation 1 was implemented using ASReml software (Gilmour et al. 2006).
Volatile networks
Correlation networks (Arkin et al. 1997; Steuer 2006) offer a way to investigate patterns of interdependency between flavour volatiles. In correlation networks, a set of nodes (volatiles) is connected by a system of edges (correlations between the volatiles). The size of the vertices (or nodes) is proportional to the absolute value of their coordinate in the first eigenvector and expresses how strongly they belong to a group. With correlation network plots, not only can clusters of volatiles be identified, but this visualisation technique also makes it possible to see how clusters of volatiles are connected. The network plots can give insight into the functional relations among volatiles, by comparing metabolic correlation networks to known biochemical pathways. The significance levels (i.e. p values) for all pair-wise correlations were converted into q values, a measure of significance in terms of the false discovery rate, using a procedure in the R with the ‘qvalue’ package (Storey and Tibshirani 2003). In this way, significance of correlations was determined, while correcting for multiple comparisons. Correlations were called significant when q < 0.05. Only the significant correlations were used in the construction of networks in R with the ‘igraph’ package (Csardi and Nepusz 2006).
Single nucleotide polymorphism (SNP) genotyping
Seedlings were genotyped using the IRSC apple 8K SNP array v1 (Chagné et al. 2012a), based on the Infinium® II technique. Genomic DNA (gDNA) was extracted from leaves using the NucleoSpin® Plant II kit (Macherey–Nagel GmbH and Co. KG, Düren, Germany) and quantified using the Quant-iT™ PicoGreen® assay (Invitrogen). Two hundred nanograms of gDNA were used as a template for the reaction, following the manufacturer’s instructions. SNP genotypes were scored using the genotyping module (version 1.8.4) of the Illumina® GenomeStudio software (Illumina Inc.). The reliability of each genotype call was measured using the GenCall score set at a minimum of 0.15, which is a lower bound for calling genotypes relative to its associated cluster. SNPs were subsequently discarded using a sequence of criteria in the following order: GenCall score at the 50 % rank (50 % GC) < 0.40; cluster separation (ClusterSep) < 0.25; more than 5 % missing calls; segregation discrepancy. BEAGLE 3.1 software (Browning and Browning 2007), with default settings, was then used for imputing missing SNP genotypes.
Marker–trait association analysis
The unified mixed linear model (MLM) approach (Yu et al. 2006) that accounts for multiple degrees of relatedness (population structure and cryptic relationships) was used:
where y is the vector of phenotypic values adjusted for fixed effects (e.g. seasonal effect, which was estimated from Eq. 1), β is an unknown vector containing fixed effects, including a SNP marker, population structure (Q) and the intercept; X is the known design matrix relating to β. All other effects are the same as in Eq. 1. To avoid spurious associations that could arise from population structure, we included principal components (PCs) derived from the genotypic data matrix (n × m) as covariates (i.e. Q matrix). The optimal number of PCs was determined by forward model selection using the Bayesian information criterion as implemented in GAPIT. In Eq. 2, each SNP was tested in turn using a t test (H 0: no additive association between the SNP and trait), and p values were obtained. Uncorrected comparison-wise p value of p < 1 × 10−3 was used to identify significant marker–trait associations. Equation 2 was fitted using GAPIT software (Lipka et al. 2012).
It is likely that polymorphisms associated with complex traits (such as most flavour volatiles) often affect multiple traits, especially those that are highly correlated. Thus, we conducted multi-trait genome-wide analysis (GWA), whereby the effects of SNPs were first divided by their corresponding standard errors to calculate signed t values. Then, a multi-trait test of the effect of a SNP was calculated as:
where \({{\mathbf{t}}_{\mathbf{i}}}\) is the vector (p × 1) of effects of ith SNP on p traits, V is the correlation matrix (p × p) among the estimated SNP effects across all SNPs. The statistic \({{\text{t}}^*}\) is distributed as a Chi-squared (\({\chi^2}\)) with p degrees of freedom (Bolormaa et al. 2013). Multi-trait GWA was conducted for each of five groups of volatiles (i.e. acetate esters, ethyl esters, alcohols, terpenes and all remaining esters), as well as for all volatile compounds together.
Mode of gene action
The nature of mode of inheritance was quantified using the ratio of dominance (d) to additive (a) effects following Eckert et al. (2009). Additive (A), partial or complete dominance (D) and over-dominance (OD) were interpreted as values in the range of [−0.50 ≤ d/a ≤ 0.50], [0.50 < |d/a| < 1.25] and [|d/a| > 1.25], respectively. Additive (a) effect was calculated as the difference between the best linear unbiased estimate (BLUE) of the effect of each homozygous class at a SNP locus (\(2a\; = \;\left| {{\mu_{AA}} - \;{\mu_{BB}}} \right|\)). Dominance (d) was calculated as the difference between BLUE of the effect of the heterozygous class (AB) and the average BLUE across both homozygous classes [d = \({\mu_{AB}}\) − 0.5(\({\mu_{AA}} + \;{\mu_{BB}}\))].
Results
Traits and markers
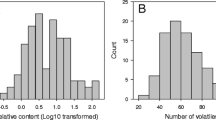
Altogether 64 volatiles were measured by GC–MS analysis of the headspace of the various apple fruit samples. Only those volatiles that were detected in more than 85 % of the seedlings were retained for further analysis. In addition, any volatile that showed insignificant variation between studied individuals was dropped. Summary statistics of the retained 37 flavour volatiles are shown in Table 1. At least 20 of these 37 volatiles (e.g. ethyl acetate, ethyl butanoate, 2-methylbutyl acetate, ethyl pentanoate, propyl acetate and ethyl hexanoate) have been previously reported as important contributors to flavour of widely grown apple cultivars including ‘Golden Delicious’, ‘Gala’, ‘Royal Gala’, ‘Cox’s Orange Pippin’ and ‘McIntosh’ (Young et al. 1996; Dixon and Hewett, 2000). The median concentration value for ethyl esters varied from 0.07 (ethyl propanoate) to 4.00 (ethyl butanoate), which were relatively lower than values for acetate esters: propyl acetate (0.41), butyl acetate (14.66), 2-methylbutyl acetate (8.82), pentyl acetate (1.36) and hexyl acetate (18.52). The highest median concentration was observed for E,E-farnesene (55.24) followed by butyl butanoate (53.37) (Table 1).
A set of 3521 SNPs was retained following various SNP data quality filters (e.g. GenCall score, cluster separation, missing calls and segregation discrepancy). The minor allele frequency (MAF) at these selected loci ranged from 0.02 to 0.50 with an average of 0.26. The retained SNPs were evenly spread across the apple genome, i.e. the proportion (out of 3521) of SNPs on any given linkage group (LG) was generally similar to the relative size of that LG assuming the total genome size as 742 megabases (Mb). The accuracy of imputing genotypes at the retained SNPs ranged from 0.85 to 1.00 with an average of 0.98.
Genetic parameters
Estimates of h 2 ranged from 0.09 (pentyl acetate, butyl acetate) to 0.71 (2-methylbutyl butanoate) (Table 1). In general, h 2 estimates of acetate esters were lower than those of other ester groups, alcohols and terpenes.
The average estimated correlation between pairs of flavour volatiles was 0.40 (Fig. 1; Supplementary Table 1), while near-perfect positive correlations were observed among volatiles within some groups, e.g. acetate esters, ethyl esters and the isomeric terpenes. The average correlation between SSC and all other volatiles was close to zero (−0.08), but moderate negative correlations were observed with hexanol (−0.28) and butanol (−0.25). Similarly to the correlations with SSC, there was a pattern of negative correlations between TA and other volatiles—the average correlation was −0.09 (Supplementary Table 1). Acetate esters showed the strongest negative correlation with TA (average −0.24), followed by an aromatic compound anethole (−0.19). One of the terpene isomers (Z,E-farnesene) showed the highest correlation (0.23) with TA. The observed genetic correlation between SSC and TA was 0.03, suggesting independent genetic control of these two traits. About half the 37 volatiles showed negative but insignificant correlations with sensory flavour, and TA was more strongly positively correlated with sensory flavour than SSC (0.37 vs. 0.15).

Heatmap of estimated genetic correlations between apple flavour volatiles. High positive correlations were observed among volatiles within some groups, e.g. acetate esters, ethyl esters and the isomeric terpenes. Soluble solids concentration (SSC) and titratable acidity (TA) were generally negatively correlated with volatiles. (Color figure online)
On average, the genotype-by-year interaction (G × Y) accounted for 12 % of the total phenotypic variation of various traits, which was far less than that contributed by differences between genotypes (61 %). Terpenes (Z,E-farnesene and E,E-farnesene) showed the highest year-to-year variation with G × Y accounting for about 25 % of the phenotypic variation. In contrast, G × Y contributed only about 5 % of the total phenotypic variation for acetate esters.
Cluster and network analysis
All individuals were used for cluster analysis, but only the small- and large-fruited individuals are shown in Supplementary Fig. 1 for clarity of grouping. Clustering of individuals based on their SNP profile revealed distinct group of small (<65 g, N = 40) and large (>200 g, N = 38) fruit size individuals (Supplementary Fig. 1A). Clustering based on flavour volatiles revealed that large-fruited individuals were more tightly clustered in the centre, while small-fruited individuals were scattered along PC1 (Supplementary Fig. 1B). Tight clustering of large-fruited individuals suggested relatively more homogeneity at the genetic and phenotypic levels in this group of individuals. The loadings of volatiles-based clustering suggested that acetate esters and ethyl esters were the key traits contributing to the grouping patterns of individuals.
We conducted correlation network analysis to understand the co-expression (or clustering) patterns of volatiles in small- and large-fruited individuals (Fig. 2). The known major classes of volatiles (i.e. acetate and ethyl esters, alcohols and terpenes) showed relatively tighter and more separated networks in large-fruited than in small-fruited individuals. The size of the node (i.e. circle) reflects the relative contribution (or expression) of different volatiles to the observed variability in a population. The size of each node is also proportional to the number of links it has, suggesting fewer significant links between volatiles in the large-fruited individuals. A relatively higher network density (0.68 vs. 0.31) observed in small-fruited individuals suggested more complex biosynthesis of volatiles than in large-fruited population (Fig. 2). The average path length (using the shortest path between each pair of nodes) and the average degree (i.e. the average number of adjacent edges of a node) of small- and large-fruited individuals were 1.32 versus 1.85, and 25 versus 12, respectively, supporting these assertions. The patterns of volatiles networks derived from all samples (N = 200) resembled to those observed in small- and large-fruited individuals (Fig. 2).

Correlation networks of volatiles in small fruit size (<65 g), large fruit size (>200 g) and all apple genotypes. Nodes represent volatiles, lines their pair-wise correlations. The size of each node (i.e. circle) reflects the relative contribution of different volatiles to the observed variability in a population and is proportional to the number of links it has. Only significant (q < 0.05) correlations are shown. Different groups of volatiles are shown in different colours: acetate esters (white), alcohol (pink), aromatic (red), ethyl esters (purple), terpenes (yellow) and other esters (green). Identification of individual volatiles is by numbers as listed in Table 1. (Color figure online)
Marker–trait associations and mode of inheritance
Acetate esters
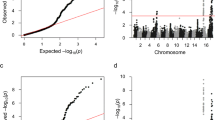
Significant associations were found between acetate esters and SNPs on LGs 2, 4 and 8 (Fig. 3a). A SNP (ss475876985) located 1979 kilobase (kb) from the top of LG2 and lying within ±50 kb of the MpAAT1 gene was associated with 2-methylbutyl acetate (2-MBA), and the second most significant SNP on LG2 (snpCO903605) showed association with pentyl acetate. GDsnp00388 (ss475882758) on LG4 showed significant association with pentyl acetate, while a SNP (ss475878971) located on LG8 showed association with some acetate esters (Fig. 3a).

Marker–trait associations for various groups of apple volatiles: acetate esters (a), ethyl esters (b), alcohols (c) and terpenes (d). Horizontal line indicates the genome-wide significance threshold (p < 0.001). Significant SNPs are indicated by the respective NCBI SNP ID (ss475xxxxxx). SNPs that were associated with volatiles in different groups are highlighted in green (bold-type). (Color figure online)
Ethyl esters
SNPs significantly associated with ethyl esters were located on LGs 1 (ss475876870), 5 (ss475878468), 10 (ss475879838) and 17 (ss475882170) (Fig. 3b). Most of these SNPs showed association with multiple ethyl esters. The second most significant SNP (GDsnp00782: ss475882287) associated with ethyl hexanoate on LG1 was in close proximity (165 kb) of a SNP within the gene MdCXE4 (MDP0000767905).
Alcohols
The two SNPs (ss475875800 and ss475876992 on LG2) showing significant association with hexanol were about 2000 kb apart, and none of these SNPs was within any annotated gene (Fig. 3c). A SNP (ss475878043) on LG4 was associated with butanol and hexanol, while a SNP (ss475878151) on LG5 was associated only with hexanol. The SNP ss475878893 on LG7 showed association with hexanol, whereas significant SNPs on LG13 (ss475880758) and LG15 (ss475881427) were associated with both hexanol and butanol (Fig. 3c). The SNP ss475880758 (LG13) is at a physical distance of about 700 kb from the gene MdLOX8b, and a significant SNP (ss475878678) on LG6 is about 340 kb away from the genes MdLOX9a. No significant association was found for 2-methylbutanol.
Terpenes
A SNP (ss475876870) on LG1, showing significant association with E,E-farnesene (Fig. 3d), also showed significant association with ethyl hexanoate (see Fig. 3b). The first and the second highly significant SNPs (ss475882675 and ss475882678, respectively) on LG2 are not within any annotated gene but are about 2000 kb away from the gene MdLOX5e. The most significant SNP associated with Z,E-farnesene on LG10 was also associated with ethyl esters. GDsnp02848 (ss475882411), which is about 700 kb away from MdLOX1e on LG12, was significantly associated with Z,E-farnesene. A SNP (ss475882170) on LG17, which showed association with ethyl esters (see Fig. 3b), was also associated with Z,E-farnesene (Fig. 3d).
No significant SNP–trait association was detected for 10 volatiles. The genomic locations of all 108 marker–trait associations identified for the rest of the 27 volatiles are shown in Supplementary Table 2. A SNP (ss475877421) on LG2, associated with multiple volatiles (hexyl 2-methylpropanoate, hexyl propanoate and butyl propanoate), is flanked by the two SNPs (ss475882675 and ss475882678) that showed association with Z,E-farnesene. The SNP (ss475878043) on LG4, showing significant association with various volatiles, is at a distance of about 600 kb from the gene MdLOX7a. A SNP (ss475878678), which is at about 340 kb distance from MdLOX9a on LG6, showed association with a number of ester and alcohol compounds (Supplementary Table 2). The SNP ss475881427 at 14.69 Mb on LG15 was associated with two alcohol compounds and six esters. In addition, the SNP ss475882170 on LG17 showed significant association with terpenes, ethyl esters and propyl 2-methylebutanoate (Supplementary Table 2).
Multi-trait GWA revealed two SNPs (ss475882758 on LG4 and ss475878971 on LG8) associated with acetate esters, while only one SNP (ss475882170 on LG17) showed significant association with ethyl esters (Supplementary Table 2). The SNP ss475875800 and ss475881427 on LG2 and LG15, respectively, were the most significant SNPs associated with alcohols. The most significant SNP on LGs 2 and 12, namely ss475882675 and ss475882411, respectively, was associated with terpenes. There were three genomic locations associated with the remaining esters: ss475876870 (LG1); ss475881427 (LG15); and ss475882170 (LG17) (Supplementary Table 2). Some SNPs were found significant in multi-trait GWA but not in single-trait analysis; for example, ss475882099 was associated with alcohols on LG17; ss475877324 and ss475878277 on LG2 and LG5, respectively, were associated with the remaining esters.
Sensory flavour intensity
The SNP array used in this study was designed to encompass SNPs in the coding region (or close proximity) of some candidate genes such as MpAAT1, MdCXE4 and MdLAR1. GDSNP00782 (ss475882287) and MdCXE4 on LG1, and MdLAR1 and MDP000294924 (ss475882883) on LGs 16 and 8, respectively, were associated with FINT (Fig. 4a) in a population of 1200 seedlings generated from crosses among six advanced selections (see Kumar et al. 2013 for details). In the germplasm accessions described here, five genomic locations, namely ss475876802 (LG1), ss475877032 (LG2), ss475878678 (LG6) and MdLAR1 and Ma (LG16), were associated with FINT (Fig. 4b). Variance explained by the significant SNPs varied between 1 and 4 %.

Genome-wide association of apple SNP markers with the sensory flavour intensity (FINT) in a breeding population (a) and germplasm (b). Titratable acidity (TA) and astringency (AST) are among the key drivers of FINT. Significant SNPs are indicated by the respective NCBI SNP ID (ss475xxxxxx). SNPs that were associated with volatiles are highlighted in green (bold-type). (Color figure online)
Mode of gene action
The putative mode of gene action at a subset of loci that were significantly associated with one or more flavour volatiles is shown in Supplementary Table 3. Majority (~85 %) of the SNP–trait associations were consistent with the additive mode of inheritance. A dominant gene action was observed at a SNP within the MpAAT1 gene associated with 2-MBA (Supplementary Table 3). Most SNP effects were small to moderate and accounted for about 3–10 % of the phenotypic variation. The number of significant marker–trait associations was the highest for hexanol, followed by ethyl hexanoate. The MAF at the significant loci varied from 0.05 to 0.47 (Supplementary Table 3), suggesting that the estimates of effect size and mode of inheritance would largely be unbiased.
Discussion
Genetic parameters
Understanding of key parameters such as the genetic control, between-trait relationships and seasonal and site variation is essential for the genetic improvement of apple flavour volatiles. The GC–MS technique has recently been used for flavour profiling of apple genotypes (Wang et al. 2007; Duan et al. 2012). Rowan et al. (2009b) reported that most of the volatiles were highly heritable (h 2 > 0.40), which is largely supported by our results here. Some of the differences in reported heritability values between the results of Rowan et al. (2009b) and our present study could be due to the size and structure of the study populations and the different growing conditions (own-rooted versus rootstock) (Kumar et al. 2011). Genotypes in our study were clonally replicated, so the high signal-to-noise ratio should provide robust heritability estimates. Apart from a few exceptions, the vast majority of all pairs of 37 volatiles were positively correlated, and near-perfect correlations (Fig. 1) were observed between volatiles within groups such as acetate esters, ethyl esters, alcohols and terpenes. These results suggest that the genetic networks responsible for the biosynthesis of apple flavour volatiles are highly interdependent. This interdependence is supported by our discovery of a number of genomic locations significantly associated with multiple volatiles.
This study demonstrated that the magnitude of G × Y interaction, across all volatiles, was one-fifth of the genotypic variation, suggesting that reliable decisions could be made about the ranking of genotypes by using only one season’s data. We obtained data only from one site, so the magnitude of genotype-by-site (G × S) interaction could not be quantified. Costa et al. (2013) reported that G × S interaction was significant only for 20 out of 181 flavour volatiles, suggesting that estimates based on one site could still be very reliable. Together with high heritabilities, largely favourable genetic correlations and minimal genotype-by-environment interactions suggested that simultaneous genetic improvement of a particular set of flavour volatiles is very possible. However, successful breeding for flavour improvement will also need an understanding of genetic relationships between flavour volatiles and sensory descriptors of apple flavour, as well as other fruit quality traits.
Volatiles networks
The average fruit weight of genotypes in our study varied between 2 and 350 g. Most commercial cultivars have an average fruit size >150 g. Primitive apple genotypes, often with small fruit sizes, are generally targeted as a source of novel traits, so the introgression plans would benefit from the understanding of volatiles network in such a gene pool. The average pair-wise correlation between all pairs of 37 volatiles in the small- and large-fruited groups was 0.46 and 0.19, respectively, and the proportion of negative correlations among all pair-wise correlations was 1 and 30 %, respectively. It appears that the simultaneous selection for larger fruit size and high eating quality (which includes sensory flavour) could have indirectly operated on a small set of flavour volatiles (especially acetate esters) so that the networks with the remaining volatiles would have been weakened. Figure 2 displays less connectivity between volatiles in the large-fruited seedling group because many of the estimated correlations were not significant. Some authors (e.g. Ursem et al. 2008; Klee and Tieman 2013) reported that the expression level and interrelationships among some flavour volatiles varied between different tomato types (e.g. cherry, beef and round) that also varied in fruit sizes.
Marker–trait associations
The chemistry of apple flavour is complex; improved flavour demands alterations of sugars, acids and volatiles synthesised through multiple parallel pathways. This chemical complexity means that multiple genes will probably need to be introduced to achieve the proper balance of flavour. The SNP array used in this study encompassed genes that are known to play roles in the biosynthesis of various esters in apple. Significant associations of SNPs residing in close proximity to genes MpAAT1 and MdCXE4 with 2-MBA and sensory flavour, respectively, provide further support for the role of these genes in imparting apple flavour. Some of the SNPs, such as ss475881427, ss475881912 and ss475882170, located on LG15, LG16 and LG17, respectively, were associated with multiple volatile esters, so these sites could be ideal for mining candidate genes for functional genomics studies. Putative QTLs identified for esters and alcohols on the top part of LG2 are consistent with those reported in previous studies (Dunemann et al. 2009; Rowan et al. 2009a; Costa et al. 2013; Souleyre et al. 2014), making this genomic location a crucial ‘hot spot’ for contributing to the biosynthesis of flavour volatiles.
Similar to some other studies (Zini et al. 2005; Dunemann et al. (2009), SNP–trait relationships were found on almost all LGs, and there were some volatiles (e.g. hexyl acetate, butyl hexanoate and 2-methylbutanol) for which no QTL was identified. In addition to a SNP (ss475876985) within ±50 kb of the MpAAT1 gene on LG2, there were two SNPs, namely ss475878971 (on LG8) and ss475882906 (on LG9), associated with 2-MBA. The per cent variation in 2-MBA explained by the SNP ss475878971 on LG8 was similar (about 6 %) to the SNP on LG2. Dunemann et al. (2009) reported a QTL for 2-MBA mapped at the top of LG9 (‘Discovery’), which is in agreement with this study. These results suggested that apart from the MpAAT1 gene, there are other genes with similar effect size contributing to the expression of 2-MBA.
Vogt et al. (2013) provide a catalogue of lipoxygenase (LOX) genes that play a decisive role in the production of apple flavour volatiles. Although SNPs from within the LOX gene family were not represented on the SNP array used in this study, a number of SNPs showing significant associations with flavour volatiles were in close proximity to the LOX genes, namely MdLOX5e (LG2), MdLOX7a (LG4), MdLOX9a (LG6), MdLOX1e (LG12) and MdLOX8b (LG13). Many important aroma and flavour volatiles are ripening-associated (Defilippi et al. 2005; Schaffer et al. 2007). Since ripening in apple is ethylene-dependent, the enzymes responsible for synthesis of some flavour volatiles could, either directly or indirectly, be driven by the transcription factors that control ethylene production (Tieman et al. 2006). The role of Md-ACO1 and Md-ACS1 genes, mapped to LG10 and LG15, respectively, in the ethylene biosynthesis is well documented (e.g. Zhu and Barritt 2008), but the physical location of significant SNPs ss475879838 (on LG10) and 475881427 (on LG15) suggested that they would be segregating independently of Md-ACO1 and Md-ACS1, respectively.
The proportion of variance explained by the significant SNPs varied between 1 and 10 %, which could partly be due to low linkage disequilibrium (LD) between SNP loci in the germplasm population used in this study. The average distance between the adjacent SNPs in our study was 170 kb, and the average LD (measured as the squared allelic correlation coefficient, r 2) between the adjacent SNPs was 0.13 (see Kumar et al. 2014). In a breeding population of highly related individuals (coefficient of relationship = 0.35), the magnitude of LD for a similar physical distance between SNPs was about 0.29 (Kumar et al. 2013). Finding SNP–trait associations in germplasm collections will require higher SNP density due to faster LD decay in such populations, so more genomic locations associated with flavour compounds can potentially be identified using higher density arrays or genotyping by sequencing. A SNP (ss475876985) linked to the AAT1 gene, explaining about 6 % of the variation in 2-MBA, was the same reported by Souleyre et al. (2014) explaining a large proportion (about 45 % on average) of variation in over 35 ester compounds in a ‘Royal Gala’ x ‘Granny Smith’ cross. Apart from potentially different LD patterns, it is also likely that this SNP could be associated with different variants of the AAT1 gene in our study. A sufficient amount of variation needs to be accounted by a marker (or two) so that it would be worthwhile for MAS to be applied. In the absence of any large-effect markers, whole genome selection would be a better tool for selection purposes (Kumar et al. 2013).
The Bayesian information criterion (BIC) was used to decide the optimal number of principal components to include as covariate to account for population structure. For all traits, the largest BIC value, indicating the goodness of fit, was obtained when no covariate was included in the model. These results suggested that the detection of SNP–trait association in this study would not be biased as there was apparently no clear population structure. The sample size (200 individuals) used here to study marker–trait associations could be considered small to identify reliable associations. However, clonal replications of individuals helped in controlling environmental noise and enhancing effective heritability. Also, the MLM approach used here reduces the number of false positives. The fact that this study confirmed some previously reported associations, suggesting that there are reliable marker–traits associations in the data presented here. For a given trait, the number of false positives in this study would be about three (= 3521 SNPs × 0.001). Following Bolormaa et al. (2010), a false discovery rate (FDR) of about 3 % is expected based on the significance threshold (p < 0.001) and the number of nominally significant SNP (108 out of 3521) in this study. Ideally, the significant SNP–trait associations should be validated in an independent data set even if a stringent threshold (or Bonferroni correction) was used to identify associations in the discovery data set.
Relationship between sensory and objective measures of flavour
Flavour is a somewhat subjective phenotype that integrates multiple sensory systems, ultimately manifesting itself at brain level. Appearance and texture certainly influence the subjective evaluation of flavour (Klee 2010), but the underlying chemistry can be reduced to molecules that interact with taste and olfactory receptors (Shepherd 2006). The relative contributions of flavour compounds (sugar, acids and volatiles) have been studied in some crops. The most significant contributor to consumer liking is sugar, with a strong positive correlation between perceived sweetness and overall liking (Klee and Tieman 2013). Volatiles can also make significant positive or negative contributions to liking. For example, the most abundant volatiles (e.g. cis-3-hexenal) in tomato do not affect consumer preferences, whereas some less abundant volatiles (e.g. geranial) do significantly affect preferences (Tieman et al. 2012).
Harker et al. (2002) showed some association between objective measures (acids, sugars, astringency) and FINT, and our results provide gene-based support for this assertion. For example, some of the genes such as Ma, MdLAR1 and MDP000294924 (ss475882883), significantly associated with FINT score (Fig. 4), were previously reported to be in association with traits such as TA and astringency (Kumar et al. 2013) and phenolics (Chagné et al. 2012b; Khan et al. 2012) which influence the sensory flavour. We also found some genomic locations (e.g. GDsnp00782, MdCXE4 and MdLOX9a) associated with FINT as well as the various flavour compounds (e.g. esters and hexanol). Phenotypic relationships of overall FINT and some descriptors (e.g. fruity and grassy) with objective measures (such as sugars, acids and esters) have been reported before, but perhaps this is the first study in apple demonstrating relationships at the molecular level. These results suggest that genomic locations of specific flavour volatiles underlying overall FINT (and flavour descriptors) can be identified from GWA studies, and these could be targeted for genetic improvement using molecular markers. We must develop selection tools to assist the breeders in maintaining and ultimately improving flavour and nutrient quality within the context of better quality and high yield.
In summary, this study showed that the majority of apple flavour volatiles are highly heritable and favourably correlated so genetic gains can be made relatively easily through conventional breeding. Effect sizes of SNP loci, some of which are associated with multiple compounds, were small (<10 %), indicating that whole genome selection, rather than MAS, is better suited for breeding for these traits. With respect to the mode of inheritance at the significant loci, the additive gene action was more common than dominant gene action. Simultaneous GWA for sensory flavour descriptors (and/or FINT) and flavour compounds is recommended for unravelling genetic mechanisms to facilitate MAS for targeted flavours in apple.
References
Arkin A, Shen P, Ross J (1997) A test case of correlation metric construction of a reaction pathway from measurements. Science 277:1275–1279
Baldwin EA, Scott JW, Einstein MA, Malundo TMM, Carr BT, Shewfelt RL, Tandon KS (1998) Relationship between sensory and instrumental analysis for tomato. J Am Soc Hort Sci 123:906–915
Bolormaa S, Pryce JE, Hayes BJ, Goddard ME (2010) Multivariate analysis of a genome-wide association study in dairy cattle. J Dairy Sci 93:3818–3833
Bolormaa S, Pyrce JE, Reverter A, Zhang YD, Barendse W, Goddard ME (2013) Multi-trait QTL mapping in beef cattle. Proc Assoc Adv Anim Breed Genet 20:515–518
Browning SR, Browning BL (2007) Rapid and accurate haplotype phasing and missing data inference for whole genome association studies using localized haplotype clustering. Am J Hum Genet 81:1084–1097
Chagné D, Crowhurst RN, Troggio M, Davey MW, Gilmore B, Lawley C, Vanderzande S, Hellens RP, Kumar S, Cestaro A, Velasco R, Main D, Rees JD, Iezzoni A, Mockler T, Wilhelm L, van de Weg E, Gardiner SE, Bassil N, Peace C (2012a) Genome-wide SNP detection, validation, and development of an 8K SNP array for apple. PLoS ONE 7:e31745
Chagné D, Krieger C, Rassam M, Sullivan M, Fraser J, André C, Pindo M, Troggio M, Gardiner SE, Henry RA, Allan AC, McGhie TK, Laing WA (2012b) QTL and candidate gene mapping for polyphenolic composition in apple fruit. BMC Plant Biol 12:12
Costa F, Cappellin L, Zini E, Patocchi A, Kellerhals M, Komjanc M, Gessler C, Biasioli F (2013) QTL validation and stability for volatile organic compounds (VOCs) in apple. Plant Sci 211:1–7
Csardi G, Nepusz T (2006) The igraph software package for complex network research. Int J Complex Syst 1695:1–9
Defilippi BG, Dandekar AM, Kader AA (2005) Relationship of ethylene biosynthesis to volatile production, related enzymes, and precursor availability in apple peel and flesh tissues. J Agric Food Chem 53:3133–3141
Dimick PS, Hoskin JC (1983) Review of apple flavour—state of the art. CRC Crit Rev Food Sci Nutr 18:387–409
Dixon J, Hewett E (2000) Factors affecting apple aroma/flavour volatile concentration: a review. NZ J Crop Hort Sci 28:155–173
Duan LL, Tian LL, Guo YR, Deng H, Li Z, Wang XY (2012) Principal component analysis and classification of aroma components of six apple cultivars. Sci Tech Food Ind 3:85–88
Dunemann F, Ulrich D, Boudichevskaia A, Grafe C, Weber WE (2009) QTL mapping of aroma compounds analyzed by headspace solid-phase microextraction gas chromatography in the apple progeny Discovery × Prima. Mol Breed 23:501–521
Dunemann F, Ulrich D, Malysheva-Otto L, Weber WE, Longhi S, Velasco R, Costa F (2012) Functional allelic diversity of the apple alcohol acyl-transferase gene MdAAT1 associated with fruit ester volatile contents in apple cultivars. Mol Breed 29:609–625
Durel C-E, Laurens F, Fouillet A, Lespinasse Y (1998) Utilisation of pedigree information to estimate genetic parameters from large, unbalanced data sets in apple. Theor Appl Genet 96:1077–1085
Eckert AJ, Bower AD, Wegrzyn JL, Pande B, Jermstad KD, Krutovsky KV, Clair JBS, Neale DB (2009) Association genetics of coastal douglas fir (Pseudotsuga menziesu var. menziesii, Pinaceae). I. Cold-hardiness related traits. Genetics 182:1289–1302
Flint J, Mackay TFC (2009) Genetic architecture of quantitative traits in flies, mice and humans. Genome Res 19:723–733
Gilmour AR, Gogel BJ, Cullis BR, Thompson R (2006) ASReml user guide release 2.0. VSN International Ltd, Hemel Hempstead
Harker FR, Marsh KB, Young H, Murray SH, Gunson FA, Walker SB (2002) Sensory interpretation of instrumental measurements 2: sweet and acid taste of apple fruit. Postharvest Biol Technol 24:241–250
Hayes BJ, Goddard ME (2001) The distribution of the effects of genes affecting quantitative traits in livestock. Genet Sel Evol 33:209–229
Karlsen AM, Aaby K, Sivertsen H, Baardseth P, Ellekjær MR (1999) Instrumental and sensory analysis of fresh Norwegian and imported apples. Food Qual Prefer 10:305–314
Khan SA, Chibon P-Y, de Vos RCH, Schipper BA, Walraven E, Beekwilder J, van Dijk T, Finkers R, Visser RGF, van de Weg EW, Bovy A, Cestaro A, Velasco R, Jacobsen E, Schouten HJ (2012) Genetic analysis of metabolites in apple fruit indicates an mQTL hotspot for phenolic compounds on linkage group 16. J Expr Bot 63:2895–2908
Klee HJ (2010) Improving the flavor of fresh fruit: genomics, biochemistry, and biotechnology. New Phytol 187:44–56
Klee HJ, Tieman DM (2013) Genetic challenges of flavour improvement in tomato. Trends Genet 29:4
Kumar SK, Volz RK, Alspach PA, Bus VGM (2010) Development of a recurrent apple breeding programme in New Zealand: a synthesis of results, and a proposed revised breeding strategy. Euphytica 173:207–222
Kumar S, Volz RK, Weskett R (2011) Genetic architecture of fruit quality traits in Malus × domestica (Borkh.) compared between own-rooted seedlings and vegetative propagules on ‘M. 9’ rootstock. Tree Genet Genomes 7:1079–1088
Kumar S, Garrick DG, Bink MCAM, Whitworth C, Chagné D, Volz RK (2013) Novel genomic approaches unravel genetic architecture of complex traits in apple. BMC Genomic 14:393
Kumar S, Raulier P, Chagné D, Whitworth C (2014) Molecular-level and trait-level differentiation between the cultivated apple (Malus × domestica Borkh.) and its main progenitor Malus sieversii. Plant Genet Resour 12:330–340
Laurens F (1999) Review of the current apple breeding programmes in the world: objectives for scion cultivar improvement. Acta Hortic 484:163–170
Li D, Xu Y, Xu G, Gu L, Li D, Shu H (2006) Molecular cloning and expression of a gene encoding alcohol acyltransferase (MdAAT2) from apple (cv. Golden Delicious). Phytochemistry 67:658–667
Lindgren D, Mullin TJ (1998) Relatedness and status number in seed orchard crops. Can J For Res 28:276–283
Lipka AE, Tian F, Wang Q, Peiffer J, Li M, Bradbury PJ, Gore MA, Buckler ES, Zhang Z (2012) GAPIT: genome association and prediction integrated tool. Bioinformatics 28:2397–2399
Mehinagic E, Royer G, Symoneaux R, Jourjon F, Prost C (2006) Characterization of odor-active volatiles in apples: influence of cultivars and maturity stage. J Agric Food Chem 54:2678–2687
Nieuwenhuizen NJ, Sa Green, Chen X, Bailleul EJD, Matich AJ, Wang MY, Atkinson RG (2013) Functional genomics reveals that a compact terpene synthase gene family can account for terpene volatile production in apple. Plant Physiol 161:787–804
Paillard NMM (1990) The flavour of apples, pears and quinces. In: Morton ID, MacLeod AJ (eds) Food flavours, Part C. The flavour of fruit. Elsevier Science Publishing Company Inc., Amsterdam, pp 1–41
Rowan DD, Hunt MB, Alspach PA, Whitworth C, Oraguzie N (2009a) Heritability, genetic and phenotypic correlations of apple fruit volatiles in a genetically diverse breeding population. J Agric Food Chem 57:7944–7952
Rowan DD, Hunt MB, Dimouro A, Alspach PA, Weskett R, Volz RK, Gardiner SE, Chagné D (2009b) Profiling fruit volatiles in the progeny of a ‘Royal Gala’ × ‘Granny Smith’ apple (Malus × domestica) cross. J Agric Food Chem 57:7953–7961
Schaffer RJ, Friel EN, Souleyre EJF, Bolitho K, Thodey K, Ledger S, Bowen JH, Ma J-H, Nain B, Cohen D, Gleave AP, Crowhurst RN, Janssen BJ, Yao J-L, Newcomb RD (2007) A genomics approach reveals that aroma production in apple is controlled by ethylene predominantly at the final step in each biosynthetic pathway. Plant Physiol 144:1899–1912
Shepherd G (2006) Smell images and the flavour system in the human brain. Nature 444:316–321
Souleyre EJF, Greenwood DR, Friel EN, Karunairetnam S, Newcomb RD (2005) An alcohol acyl transferase from apple (cv. Royal Gala), MpAAT1, produces esters involved in apple fruit flavour. FEBS J 272:3132–3144
Souleyre EJF, Marshall SDG, Oakeshott JG, Russell RJ, Plummer KM, Newcomb R (2011) Biochemical characterisation of MdCXE1, a carboxylesterase from apple that is expressed during fruit ripening. Phytochemistry 7:564–571
Souleyre EJF, Chagné D, Chen X, Tomes S, Turner RM, Wang MY, Maddumage R, Hunt MB, Winz RA, Wiedow C, Hamiaux C, Gardiner SE, Rowan DD, Atkinson RG (2014) The AAT1 locus is critical for the biosynthesis of esters contributing to ‘ripe apple’ flavour in ‘Royal Gala’ and ‘Granny Smith’ apples. Plant J 78:903–915
Steuer R (2006) On the analysis and interpretation of correlations in metabolomic data. Brief Bioinform 7:151–158
Storey JD, Tibshirani R (2003) Statistical significance for genomewide studies. Proc Natl Acad Sci USA 100:9440–9445
Tieman DM, Zeigler M, Schmelz EA, Taylor MG, Bliss P, Kirst M, Klee HJ (2006) Identification of loci affecting flavour volatile emissions in tomato fruit. J Exp Bot 57:887–896
Tieman D, Bliss P, McIntyre LM, Blandon-Ubeda A, Bies D, Odabasi AZ, Rodrıguez GR, van der Knaap E, Taylor MG, Goulet C, Mageroy MH, Snyder DJ, Colquhoun T, Moskowitz H, Clark DG, Sims C, Bartoshuk L, Klee HJ (2012) The chemical interactions underlying tomato flavor preferences. Curr Biol 22:1035–1039
Ulrich D, Dunemann F (2012) Towards the development of molecular markers for apple volatiles. Flavour Fragr J 27:286–289
Ursem R, Tikunov Y, Bovy A, Van Berloo R, Van Eeuwijk F (2008) A correlation network approach to metabolic data analysis for tomato fruit. Euphytica 161:181–193
Vogt J, Schiller D, Ulrich D, Schwab W, Dunemann F (2013) Identification of lipoxygenase (LOX) genes putatively involved in fruit flavour formation in apple (Malus × domestica). Tree Genet Genomes 9:1493–1511
Wang HB, Chen XS, Xin PG, Feng T, Shi J, Ci ZJ (2007) GC–MS analysis of volatile components in several early apple cultivars. J Fruit Sci 1:003
Young H, Gilbert JM, Murray SH, Ball RD (1996) Causal effects of aroma compounds on Royal Gala apple flavours. J Sci Food Agri 71:329–336
Yu J, Pressoir G, Briggs WH, Vroh-Bi I, Yamasaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen DM, Holland JB, Kresovich S, Buckler ES (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208
Zhu Y, Barritt BH (2008) Md-ACS1 and Md-ACO1 genotyping of apple (Malus × domestica Borkh.) breeding parents and suitability for marker-assisted selection. Tree Genet Genomes 4:555–562
Zhu Y, Rudell DR, Mattheis JP (2008) Characterization of cultivar differences in alcohol acyltransferase and 1-aminocyclopropane-1-carboxylate synthase gene expression and volatile ester emission during apple fruit maturation and ripening. Postharvest Biol Technol 49:330–339
Zini E, Biasioli F, Gasperi F, Mott D, Aprea E, Mark T, Patocchi A, Gessler C, Komjanc M (2005) QTL mapping of volatile compounds in ripe apples detected by proton transfer reaction-mass spectrometry. Euphytica 145:269–279
Acknowledgments
The PFR Markers and Mapping Team is thanked for help in collecting leaf samples. Slipstream Automation Ltd, New Zealand, is thanked for providing high-throughput DNA extraction services. We also thank AgResearch Invermay, New Zealand, for providing Illumina® SNP array genotyping. This research was partly supported by the New Zealand Ministry of Business, Innovation and Employment (MBIE). PFR colleagues Richard Volz and Nigel Perry are thanked for their constructive feedback.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kumar, S., Rowan, D., Hunt, M. et al. Genome-wide scans reveal genetic architecture of apple flavour volatiles. Mol Breeding 35, 118 (2015). https://doi.org/10.1007/s11032-015-0312-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-015-0312-7