
Abstract
Mango (Mangifera indica) is the most important horticultural fruit crop in India, but few genetic markers have been identified in it. In order to develop genomic marker resources for mango, we sequenced genomic DNA using next-generation sequencing technology on the IlluminaHiSeq 2000 platform and examined sequence data for microsatellite markers. High-quality raw data were assembled and 198,612 contigs were obtained after optimization. From these data, 159,228 scaffolds were generated covering a genome size of 253.6 Mbp. From the scaffolds, 106,049 microsatellite repeats were identified. Finally, we were able to design primers for 84,118 microsatellites. Ninety simple sequence repeat (SSR) markers were tested, employing 64 mango cultivars and four Mangifera species, for determination of polymorphism and cross-species amplification. We identified 2103 alleles, and the allele number per locus ranged from 15 to 36. The majority of these markers amplified DNA in related species with a transferability of 94.4–98.8 %. The present study increases the sequence coverage of the mango genome and the number of mango-specific SSR markers. This is also the first report of the development of genomic SSR markers in mango using next-generation sequence technology. The genomic SSR markers identified in this study will be useful in diversity, identification, mapping and breeding studies.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Mango (Mangifera indica) is a dicotyledonous fruit tree of the order Sapindales, family Anacardiaceae, with chromosome number 2n = 40 and genome size of 4.39 × 108 bp (Arumuganathan and Earle 1991). Mango is a cross-pollinated, nutritionally important fruit, rich in minerals and vitamins, and is widely grown in tropical and subtropical regions. Genetic information in mango is limited owing to limited systematic breeding efforts and poor utilization of wild mango species. The situation is worsened by a lack of genomic information. Identification of molecular markers tightly linked to the important agronomic traits is necessary for undertaking molecular breeding in plants (Ratnaparkhe et al. 1995; Yang et al. 2006; Odeny et al. 2009; Dutta et al. 2011; Bohra et al. 2011). Here we report identification and characterization of microsatellite markers in M. indica. This provides a platform to accelerate future improvement of this economically important tree fruit crop.
Materials and methods
Plant materials
Genomic DNA from the mango cultivar Alphonso was used for genomic sequencing and identification of simple sequence repeat (SSR) markers. To characterize SSR markers and determine cross-species transferability, we employed 64 cultivars and four related species, M. anadamanica, M. camptosperma, M. odorata and M. griffthi (Electronic Supplementary Material ESM-1). The plant material was obtained from the germplasm collection of ICAR—Indian Institute of Horticultural Research, Hessarghatta Lake Post, Bengaluru, India.
Genome sequencing and assembly
High-quality genomic DNA was isolated from leaves of cultivars and species using a modified CTAB method (Ravishankar et al. 2000). DNA (25–50 ng) from the cv. Alphonso was used to generate genomic sequences. Genomic DNA was sequenced using an Illumina HiSeq 2000 platform at M/s Biokart, Bangalore facility, following the manufacturer’s instructions (www.Illumina.com). The raw data obtained were examined using the Trueseq Library (paired-end). High-quality (HQ) data were used for analysis [HQ bases: bases having Phred score ≥20; HQ reads: reads having ≥70 % HQ bases (i.e. Phred score ≥20)]. Data were then assembled into contigs using the de Bruijn graph-based assembler Velvet (v1.1.08) software (k-mer 55; file type: Fastq; read type: Short Paired; Daniel and Ewan 2008). The large sequence contigs were quality checked and contaminating sequences were identified and removed. Assembly with k-mer 55 was selected for scaffolding as it has the optimal readings for N50, percent assembly and number of contigs generated (ESM2). Later scaffolds were generated from pre-assembled contigs (SSPACE basic software v1.0; Boetzer et al. 2011).
Survey, identification and primer design for genomic SSR markers
All assembled scaffolds were screened for the presence of SSRs using MISA software (http://pgrc.ipk-gatersleban.de/misa). MISA files were transferred to Microsoft Excel where SSRs were classified into mono-, di-, tri-, tetra-, penta- and hexa-nucleotide and compound repeats. Primer pairs flanking the repeats were designed using Primer3 software (http://wwwgenome.wi.mit.edu/genome_software/other/Primer3.htm; Steve and Skaletsky 1996) with the following settings: product size: 100–280 bp; primer melting temperatures: min 57 °C, optimum 60 °C, max 63 °C; primer size: min 18 bp, optimum 20 bp and max 27 bp. Redundancy analysis of primer sequences was carried out for forward and reverse primers separately, using Microsoft Excel software.
PCR and genotyping
Genomic DNA of 64 mango genotypes was adjusted to a final concentration of 25–50 ng/µl each. A total of 110 SSR primers was randomly selected and synthesized with M13 tailed primers (17 bp forward: GTAAAACGACGGCCAGT; 7 bp reverse: GTTTCTT). These M13 tailed primers were first screened for their ability to produce amplification in mango DNA using DNA from cultivars Alphonso and Neelum. We used the fluorescence-based PCR method of Schuelke (2000) to amplify the microsatellites in a quick, accurate and efficient manner. PCR was carried out in a 15-µl reaction volume containing 1.5 µl of reaction buffer (pH 9.0, 10 mM Tris with 15 mM MgCl2, 50 mM KCl and 0.01 % gelatin), 0.3 µl of 25 mM MgCl2, 1.5 µl of 1 mM dNTPs, 0.8 µl (5 pmol) of forward primer, 0.8 µl of reverse primer (5 pmol), 0.8 µl of labeled M13 probe (5 pmol; FAM, VIC, NED and PET), 3.0 µl (75 ng) of template genomic DNA, 0.54 µl (3 U/µl) of Taq DNA polymerase and 5.8 µl of nuclease-free water. The PCR cycling profile was an initial denaturation at 94 °C for 2 min, followed by 35 cycles of 94 °C for 30 s, 55 °C for 30 s and 72 °C for 1 min, and a final extension at 72 °C for 5 min. PCR reaction was carried out using an Eppendorf Master Cycler Gradient thermocycler (Eppendorf AG, Hamburg, Germany). Amplified products were initially separated on 3 % agarose gel for confirmation of the amplification. To reduce the cost of genotyping, samples were mixed by combining four PCR products labeled with different fluorophores (FAM, VIC, NED and PET) into a single sample. These samples were separated on an automatic 96-capillary automated DNA sequencer (ABI 3730 DNA Analyzer, Applied Biosystems, USA) at ICRISAT facility, Hyderabad, India.
Genetic analysis of SSR markers
The raw data generated was analyzed and compiled using Peak Scanner v1.0 software (Applied Biosystems) to determine allele sizes. The results obtained were used for genetic analysis with Cervus 3.0 software (Kalinowski et al. 2007). We estimated the number of alleles, observed heterozygosity (Ho), expected heterozygosity (He) and polymorphic information content (PIC) using Cervus 3.0 software.
Results
Sequence analysis, assembly, SSR identification and primer design
The next-generation sequencing (NGS) technology Illumina HiSeq 2000 platform was used to sequence genomic DNA from cv. Alphonso. The sequencing run yielded 9,122,660,471 bases from 90,323,371 reads. Low-quality reads were filtered out. Finally, 172.92 million (95.72 %) paired-end reads were obtained. Assembly optimization was accomplished using Velvet software (Daniel and Ewan 2008). Assembly with k-mer 55 was selected, as it has the optimal reading for N50 (ESM 2). This resulted in 198,612 contigs with total assembly size 253,758,328 bases. Contig size ranged from 300 to 44,106 bp. Later, genome assembly was done using the contigs and this generated 159,228 scaffolds. The shortest scaffold length was 300 and the longest 71,146 bp. The weighted mean assembly size in scaffold (N50) was 3249 bp. The total assembled size of the scaffold was 253.65 Mbp (Table 1).
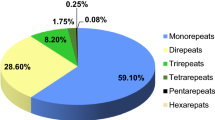
An SSR survey of the genomic sequences using MISA software (http://pgrc.ipk-gatersleban.de/misa) revealed that 66,288 scaffolds contained 106,049 SSRs, of which 84,118 were usable. Primers were designed for these SSRs (ESM 3); 21,931 SSRs were unsuitable for primer design, accounting for 20.6 % of the total identified SSRs. The redundancy analysis of primer sequences showed that 185 and 1510 were redundant for forward and reverse primers, respectively, amounting to redundancy of 0.2 % for forward primers and 1.79 % for reverse primers. Among the SSR repeats identified, mono-nucleotide repeats were the most abundant, accounting for 52.9 % of total SSRs, followed by di-nucleotide repeats (14.9 %), tri-nucleotide repeats (15.9 %), tetra-nucleotide repeats (3.5 %), penta-nucleotide repeats (1.1 %) and hexa-nucleotide repeats (0.47 %) and compound nucleotide repeat types (11.3 %) (Table 2). Among the di-nucleotide repeat motifs, the AT and TA repeats were the most common, while the predominant tri-nucleotide motifs were AAT and TTA (Table 3). AT-rich repeats were also the most common repeats in tetra-nucleotide, penta-nucleotide and hexa-nucleotide SSRs.
Genetic analysis and transferability of genomic SSRs
Genetic analysis of 90 SSR markers showed PIC values ranging from 0.738 to 0.960 with a mean of 0.8999. The values of observed and expected heterozygosity ranged from 0.00 to 0.177 and from 0.753 to 0.969, respectively. The number of alleles per locus ranged from 15 to 36 with a mean of 23.37 (Tables 4, 5). Transferability of the 90 genomic SSR markers was tested using DNA of four other species: M. andamanica, M. camptosperma, M. odorata and M. griffthi. A high rate of transferability was observed for these species (ESM 3): 87 SSR markers amplified PCR products in M. andamanica (96.6 %), 89 in M. griffthi (98.8 % transferability), 87 in M. odorata (96.6 % transferability) and 85 in M. camptosperma (94.4 % transferability; ESM4).
Discussion
Genomic SSRs markers are widely used for mapping in crop breeding programs, and in population genetics, because they are easy to amplify, reliable, polymorphic and often multi-allelic (Varshney et al. 2005). Apart from these desirable characteristics, they are also easily amenable to automation. However, the use of microsatellite markers for studying non-model species like mango has been impeded by lack of available genomic resources. Unfortunately, until now only 108 SSR markers for mangos have been published (Ravishankar et al. 2011, Schnell et al. 2005; Viruel et al. 2005; Duval et al. 2005; Honsho et al. 2005; Chiang et al. 2012; Ukoskit 2007). Previously, detection of genomic SSRs and their subsequent establishment as markers was expensive and time-consuming, involving construction and screening of microsatellite-enriched genomic DNA libraries (Glenn and Schable 2005). Compared to this method using probes, which is widely used for identifying microsatellites from species with little genetic information, the present NGS-based method is fast and simple, and overcomes a number of technical difficulties. The advent of NGS technologies has made this process easier and less complicated (Zalapa et al. 2012). Much of the work is now performed in silico with wet laboratory experiments confined to SSR validation. As a result, large numbers of SSR markers can be developed in a short time span and at lower cost. This approach is especially useful for many tree crops where there are no or only a few SSR markers available.
Distribution of SSRs in the mango genome
We used the high-throughput sequencing platform to identify genomic SSR markers in mango. The assembly of reads of the long sequences (average length 1593 bp) resulted in 159,228 scaffolds covering 253 Mb of the mango genome (Table 1). This is approximately 57.6 % of the mango genome, which has been reported to contain 439 Mbp of DNA (Arumuganathan and Earle 1991). We observed that mono-nucleotide repeats are the predominant type of repeat in mango and accounted for 52.9 % of observed repeats, followed by tri-nucleotide (15.9 %) and di-nucleotide (14.9 %) repeats (Table 2).
Mono-nucleotide repeats are the most common type of repeats in monocot (rice, sorghum and Brachypodium) and also in dicot (Arabidopsis, Medicago and Populus) species. Mono-nucleotide repeats were found to be lowest in sorghum (43 %) and highest in Medicago (79 %) genomes (Sonah et al. 2011). We found that the mono-, di- and tri-nucleotide repeats contribute to the major proportion of SSRs in mango and a very small portion was contributed by tetra-, penta- and hexa-nucleotide repeats (Table 2).
Tri-nucleotide repeats are also the most frequently identified SSR type in many plant species, such Arabidopsis, rice, soybean and sorghum genomic DNA (Cavagnaro et al. 2010; Sonah et al. 2011). The abundance of tri-nucleotides in genomic DNA is hypothesized to be the result of purifying selection which eliminates any SSRs causing frame-shift mutations. However, it is unknown whether selection is involved in the distribution of SSR motifs (Celik et al. 2014). The average density of genomic SSRs was one SSR for every 4.2 kb of genomic DNA, which is within the range observed in many plant species (Cavagnaro et al. 2010).
Of the different motifs, AT/TA-rich motifs were often the most common (Table 3). This pattern is similar in genomic DNA of other dicot plant species (Cavagnaro et al. 2010; Selale et al. 2013). We found that AAT/TTA was the most common tri-nucleotide motif, which is in agreement with results reported for other dicots, Medicago and Populus (Sonah et al. 2011).
The dominant occurrence of a particular repeat motif and its length in the plant genome is the outcome of selection pressure applied to that specific motif during evolution. However, the molecular mechanism of the origin and evolution of microsatellites is not clearly understood. The most common mutational mechanism assumed to be operating is replication slippage, by which addition or removal of one or more motifs might have happened. The other processes, like unequal crossing over, nucleotide substitution, and duplication events, may also be responsible for microsatellite variation. However, they may not explain species-specific accumulation of particular motif repeats (Sonah et al. 2011; Buschiazzo and Gemmell 2006).
Genetic analysis and transferability of genomic SSR markers
In this study, only 90 SSR primers from initially selected 110 primers amplified products in PCR for mango. A high rate of successful amplification can be due to high-quality sequence data and the appropriate primer parameters, such as high GC content. In our study, the genomic SSR markers detected a high level of polymorphism, with an average PIC value of 0.8999, which is higher than values reported in previous studies (Ravishankar et al. 2011; Chiang et al. 2012; Schnell et al. 2005). Genomic SSRs are often reported to have higher levels of polymorphism than genic SSRs (Varshney et al. 2005). All the SSR markers in this study amplified multiple alleles (average of 23.37 per locus; Tables 4, 5). The average number of alleles per locus is also higher than in previous studies on mango by Schnell et al. (2005), Honsho et al. (2005), Viruel et al. (2005) and Duval et al. (2005). This may be due to the diverse and large number of genotypes used in this study, which helped to capture a large number of alleles. In our study, 64 SSR markers (71 %) had more than 20 alleles per locus, indicating high heterozygosity and diversity of cultivars used (Table 4).
High transferability of the genomic SSR markers to related Mangifera species was observed, varying from 94.4 to 98.8 % depending on the species tested. High transferability has also been reported for SSR markers in Papaver species (Celik et al. 2014; Selale et al. 2013). The high level of transferability indicates that there are highly conserved regions in the flanking sequences of microsatellites among Mangifera species.
The next-generation sequencing and mining of the mango genome helped in identification of 84,118 SSR loci. Ninety SSR markers standardized for mango showed high PIC values and have shown high transferability within the genus Mangifera. The information generated in this study will form an important repertoire of molecular tools for genetic studies and breeding in mango.
References
Arumuganathan K, Earle ED (1991) Estimation of nuclear DNA content of plants by flow cytometry. Plant Mol Biol Report 9:221–231
Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W (2011) Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27(4):578–579
Bohra A, Dubey A, Saxena RK, Penmetsa RV, Poornima KN (2011) Analysis of BAC-end sequences (BESs) and development of BES-SSR markers for genetic mapping and hybrid purity assessment development in pigeonpea (Cajanus spp.). BMC Plant Biol 11:56
Buschiazzo E, Gemmell NJ (2006) The rise, fall and renaissance of microsatellites in eukaryotic genomes. Bioessays 28(10):1040–1050
Cavagnaro PF, Senalik DA, Yang L, Simon PW, Harkins TT, Kodira CD, Huang S, Weng Y (2010) Genome-wide characterization of simple sequence repeats in cucumber (Cucumis sativus L.). BMC Genom 11:569–586
Celik I, Gultekin V, Allmer J, Doganlar S, Frary A (2014) Development of genomic simple sequence repeat markers in opium poppy by next-generation sequencing. Mol Breed 34:323–334
Chiang YC, Tsai CM, Chen YKH, Lee SR, Chen CH, Lin YS, Tsai CC (2012) Development and characterization of 20 new polymorphic microsatellite markers from Mangifera indica (Anacardiaceae). Am J Bot 99:e117–e119
Daniel RZ, Ewan B (2008) VELVET: algorithm for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829. doi:10.1101/gr.074492.107
Dutta S, Kumawat G, Singh BP, Gupta DK, Singh S, Dogra V, Gaikwad K, Sharma TR, Raje RS, Bandhopadhya TK, Datta S, Singh MN, Bashasab F, Kulwal P, Wanjari KB, Varshney RK, Cook DR, Singh NK (2011) Development of genic-SSR markers by deep transcriptome sequencing in pigeonpea [Cajanus cajan (L.) Millspaugh]. BMC Plant Biol 11:17
Duval MF, Bunel J, Sitbon C, Risterucci AM (2005) Development of microsatellite markers of mango. Mol Ecol Notes 5:824–826
Glenn TC, Schable TC (2005) Isolating microsatellite DNA loci. Methods Enzymol 395:202–222
Honsho C, Nishiyama K, Eiadthong W, Yonemori K (2005) Isolation and characterization of new microsatellites markers in mango. Mol Ecol Notes 5:152–154
Kalinowski ST, Taper ML, Marshall TC (2007) Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol Ecol 16:1099–1106
Odeny DA, Jayashree B, Gebhardt Crouch J (2009) New microsatellite markers for pigeonpea (Cajanus cajan (L.) Millsp.). BMC Res Notes 2:35. doi:10.1186/1756-0500-2-35
Ratnaparkhe MB, Gupta VS, Murthy MRV, Ranjekar PK (1995) Genetic fingerprinting of pigeonpea [Cajanus cajan (L.) Millsp.] and wild relatives using RAPD markers. Theor Appl Genet 91:893–898
Ravishankar KV, Anand L, Dinesh MR (2000) Assessment of genetic relatedness among a few Indian mango cultivars using RAPD markers. J Hortic Sci Biotechnol 75:198–201
Ravishankar KV, Mani BH, Anand L, Dinesh MR (2011) Development of new microsatellite markers from mango (Mangifera indica) and cross-species amplification. Am J Bot 98(4):e96–e99
Schnell RJ, Olano CT, Quintanilla WE, Meerow AW (2005) Isolation and characterization of 15 microsatellite loci from mango (Mangifera indica L.) and cross-species amplification in closely related taxa. Mol Ecol Notes 5:625–627
Schuelke M (2000) An economic method for the fluorescent labelling of PCR fragments. Nat Biotechnol 18:233–234
Selale H, Celik I, Gultekin V, Allmer J, Doganlar S, Frary A (2013) Development of EST-SSR markers for diversity and breeding studies in opium poppy (Papaver somniferum L.). Plant Breed 132:344–351. doi:10.1111/pbr.12059
Sonah H, Deshmukh RK, Sharma A, Sing VP, Gupta DK, Gacche RN, Rana JC, Singh NK, Sharma TR (2011) Genome-wide distribution and organization of microsatellites in plants: an insight into marker development in Brachypodium. PLoS ONE 6(6):e21298:1–e21298:9
Steve R, Skaletsky HJ (1996) Primer 3. http://wwwgenome.wi.mit.edu/genome_software/other/Primer3.html
Ukoskit K (2007) Development of microsatellite markers in mango (Mangifera indica L.) using 5′ anchored PCR. Thammasat Int J Sci Technol 12:3
Varshney RK, Graner A, Sorrells ME (2005) Genic microsatellites markers in plants: features and application. Trends Biotechnol 23:48–55
Viruel MA, Escribano P, Barbieri M, Ferri M, Hormaza JI (2005) Fingerprinting, embryo type and geographic differentiation in mango (Mangifera indica L., Anacardiaceae) with microsatellites. Mol Breed 15:383–393. doi:10.1007/s11032-004-7982-x
Yang SY, Pang W, Ash G, Harper J, Carling J, Wenzl P, Huttner E, Zong XX, Kilian A (2006) Low level of genetic diversity in cultivated pigeon pea compared to its wild relatives is revealed by diversity arrays technology. Theor Appl Genet 113:585–595
Zalapa JE, Cuevas H, Zhu H, Steffan S, Senalik D, Zeldin E, McCown B, Harbut R, Simon P (2012) Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am J Bot 99:193–208
Acknowledgments
K.V.R. and M.R.D. acknowledge financial support from Department of Biotechnology, New Delhi funded projects “Morphological and Molecular Characterization of Wild and Indigenous Mango Varieties of Indo-Burma Region (North-Eastern Region) and National Database on Mango”.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ravishankar, K.V., Dinesh, M.R., Nischita, P. et al. Development and characterization of microsatellite markers in mango (Mangifera indica) using next-generation sequencing technology and their transferability across species. Mol Breeding 35, 93 (2015). https://doi.org/10.1007/s11032-015-0289-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-015-0289-2