
Abstract
Next-generation sequencing (NGS) technologies open up a wealth of opportunities for plant breeding and genomic research and change the paradigms of DNA marker detection, genotyping, and gene discovery. Abundant genomic resources have been generated using a whole-genome resequencing (WGR) strategy and utilized in genome-wide association, genome diversity, and evolution studies in many crops with a reference genome such as rice and maize. The WGR-based quantitative trait loci mapping approach developed in soybean combines single nucleotide polymorphism (SNP) discovery, validation and genotyping and has the potential to identify candidate genes and causal SNPs without a time-consuming fine-mapping process. Given that this approach solves issues caused by genome duplications and repetitive sequences, it can be widely utilized in crops with a reference genome. The combination of WGR with bulked segregant analysis provides a rapid way to identify genes or causal mutations. Currently, DNA sequencing technologies are being improved rapidly. Third-generation sequencing platforms can overcome some inherent disadvantages of NGS and are expected to promote the application of WGR-based approaches and revolutionize plant breeding, genomic and genetic research.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A central task of modern plant genetics and breeding is to understand the genetic basis of phenotypic variations. Since Lander and Botstein (1989) published their milestone paper, an avalanche of molecular mapping studies ensued, and tremendous resources have been directed to mapping quantitative trait loci (QTL) for traits of economical importance using DNA markers. However, traditional molecular mapping is a time-consuming, expensive, and laborious procedure involving complicated marker discovery (DNA fragment cloning, sequencing, primer design, and marker validation) and both genotyping and phenotyping of mapping populations. Thus, although a large number of genes/QTL have been mapped in major crops (Bernardo 2008), the progress in cloning genes and identifying causal quantitative trait nucleotides (QTN) for important traits is slow mainly because of low mapping resolution. Most map-based cloning projects have ended up with identifying a large genomic region containing many genes. Identification of causative genes and markers is imperative for improving the accuracy of marker-assisted selection. Thus, more efficient QTL mapping and cloning approaches are needed.
Advances in next-generation sequencing (NGS) technologies have not only greatly facilitated de novo sequencing of crop genomes, but also opened up a wealth of previously impossible opportunities for plant breeding and genetic studies. Given that raw sequencing output is doubling roughly every 6 months and NGS costs continue falling (Poland and Rife 2012), NGS technologies are routinely utilized to detect sequence variations (Deschamps et al. 2010; Hyten et al. 2010; Van Orsouw et al. 2007; Baird et al. 2008), and map QTLs or genes of agronomic importance (Andolfatto et al. 2011; Huang et al. 2009). Whole-genome resequencing (WGR) of plant genomes allows the discovery of a huge number of DNA markers such as SNPs, Indels, copy number variations (CNV), and presence/absence variations (PAV) in crops and provides deep insight into genome evolution (Huang et al. 2010, 2012a, b; Jiao et al. 2012; Lai et al. 2010; Chia et al. 2012; Lam et al. 2010; Xu et al. 2012). WGR-based approaches are revolutionizing QTL mapping by augmenting accuracy, output, speed, and cost-effectiveness of genome-wide genotyping (Huang et al. 2009; Xu et al. 2013). Moreover, the combination of WGR with bulked segregant analysis (BSA) allows rapid identification of genes and causal mutations in crops with a reference genome (Abe et al. 2012; Takagi et al. 2013a, b), and the application of NGS technologies in targeting induced local lesions in genomes (TILLING) provides a robust approach to discover rare mutations in populations (Tsai et al. 2011). In the past few years, a number of WGR-based approaches have been emerged to exploit the full potential of DNA sequencing. This review article discusses the progress in use of these methods in crop genomic and genetic research.
WGR simplifies marker discovery
Molecular markers lie at the heart of modern plant genetics research. Large-scale maker discoveries are started with the landmark decoding of the Arabidopsis thaliana cv. Columbia (Col-O) genome (The Arabidopsis Genome Initiative 2000). With a high-quality Col-O reference genome sequence, mainly assembled using a BAC-by-BAC approach, plant geneticists were able to identify a large set of SNPs and Indels by resequencing another strain, Landsberg erecta (Ler), at a low coverage (2×) using a shotgun approach. In rice, the genome sequences of Nipponbare and 93-11 (International Rice Genome 2005; Goff et al. 2002; Yu et al. 2002; Feltus et al. 2004) were manipulated in the same way as those of Col-O and Ler in A. thaliana. These genomic resources greatly improved the efficiency of gene and QTL mapping and cloning in these species.
The advent of NGS technologies revolutionized the way we detect DNA markers. A reduced representation sequencing (RRS) strategy based on properties of restriction enzymes has been widely utilized in SNP discovery. Similar to the principal of amplified fragment length polymorphism marker, RRS uses one or two restriction enzymes to digest genomic DNA, subsequently sequences a subset of restriction fragments, and produces partial, but genome-wide, coverage at a low cost. Polymorphisms in the resulting fragments among genotypes can be used as molecular markers. Protocols based on this strategy include reduced representation library and complexity reduction of polymorphic sequences (Van Tassell et al. 2008), restriction-site-associated DNA tag (Baird et al. 2008), multiplexed shotgun genotyping (MSG) (Andolfatto et al. 2011), and genotyping-by-sequencing (GBS) (Elshire et al. 2011). These RRS approaches are the method of choice for crops such as wheat that have no reference genome sequence available (Poland et al. 2012a, b), and are also valuable for crops with a complex or large genome because researchers can target low copy sequences by choosing methylation-sensitive enzymes for digestion to avoid cutting methylated transposons (Elshire et al. 2011).
An alternative approach is WGR. Advances in NGS technologies have rapidly increased sequencing output and dramatically brought down sequencing costs. As a result, reference genome sequences are available for many crops including rice (International Rice Genome 2005; Goff et al. 2002; Yu et al. 2002), maize (Schnable et al. 2009), soybean (Schmutz et al. 2010), barley (Mayer et al. 2012), potato (Potato Genome Sequencing Consortium 2011), tomato (Tomato Genome Consortium 2012), and cotton (Li et al. 2014). With the availability of these high-quality reference genome sequences, resequencing a panel of diverse germplasm becomes routine for studying genome diversity, identifying selection signatures, and discovering sequence variations. This strategy was widely used in rice (Huang et al. 2010, 2012a, b; Xu et al. 2012), maize (Jiao et al. 2012; Lai et al. 2010; Chia et al. 2012), soybean (Lam et al. 2010), pigeon pea (Varshney et al. 2012), chickpea (Varshney et al. 2013), common bean (Schmutz et al. 2014), and other crops.
In addition to SNPs and Indels, the WGR approach allows the detection of structure variations such as CNVs and PAVs. Lai et al. (2010) resequenced five maize inbred lines and detected several hundreds of complete PAV genes that might contribute to maize heterosis. In another study, Chia et al. (2012) resequenced 103 cultivated and wild maize inbred lines and identified a large number of CNVs and PAVs. Structure variations are of agronomic importance. One example is the Rhg1 gene encoding resistance to soybean cyst nematode (SCN), the most economically damaging pest of soybean in the USA. Cook et al. (2012) cloned this gene and found that the SCN resistance mediated by Rhg1 is conferred by CNV of a 31-kilobase segment: One copy of this segment is present in susceptible varieties, and there are 10 tandem copies in resistant cultivars. Further study of associations between agronomic traits and structure variations will provide insights into the genetic bases of phenotypic variations and accelerate plant breeding progress.
RRS strategy for map construction
The RRS strategy is utilized to genotype mapping populations. Andolfatto et al. (2011) developed the MSG approach to genotype a mapping population of Drosophila simulans, a model species with a small genome of approximately 130 Mb. They used restriction enzyme MseI to reduce the complexity of genome sequence and implemented a hidden Markov model (HMM) to genotype each individual and determine the recombination break points. The mapping results suggested that MSG is a highly effective genotyping platform for D. simulans. However, the RRS-based approaches only sequence a limited portion of the genome. Thus, challenges may arise when this or a similar RRS-based approach is used to determine recombination break points in crops with a complex genome such as maize. Sequencing a small portion of a genome may be insufficient to identify all recombination events, and the mapping resolution may also be substantially compromised.
Another RRS-based approach, GBS, has been widely used in molecular mapping and genomic selection (Elshire et al. 2011; Poland et al. 2012b, 2013). Since reference genome sequences are not a prerequisite for GBS analysis, GBS is particularly useful for those crops without a reference genome sequence such as wheat (Poland et al. 2012b). The cost of GBS is low because only a selected portion of genomes is sequenced. Researchers also have the flexibility of sequencing genomic regions of interest by choosing different restriction enzymes (Poland et al. 2012a). However, unequal recovery of restriction fragments among samples in multiplexed sequencing results in a considerable amount of missing data, which need to be imputed with sophisticated bioinformatics tools. Given that more crop reference sequences are available and sequencing costs are falling, it is expected that RRS-based genotyping approaches will eventually phase out and be replaced by WGR in the near future.
WGR, an efficient genotyping approach for linkage mapping in rice
The WGR approach was first employed to genotype biparental mapping populations in rice. Huang et al. (2009) developed a WGR method to genotype a recombinant inbred line (RIL) population derived from two cultivars with a sequenced genome, cv. 93-11 and Nipponbare. With a bar-coded multiplexed sequencing approach, each RIL was sequenced at 0.02× coverage to identify recombinant break points. Considering that NGS is error-prone and the sequence coverage was extremely low in this study, they developed a sliding widow approach (Table 1) to collectively determine genotypes, and successfully mapped a QTL for plant height to a 100-kb region containing the rice “green revolution gene.” The power of this WGR method was further demonstrated by mapping 10 QTL for domestication-related traits to <200 kb away from the known genes (Huang et al. 2012b). More recently, this approach was adopted to dissect yield-associated loci in super hybrid rice (Gao et al. 2013). In this study, two parental lines of a RIL population, PA64s and 93-11, were sequenced at 48 and 36× depths, respectively, and each RIL at about 4× depth. Although the population size (132) was relatively small, a total of 43 QTL for yield-associated traits were mapped with a reasonable resolution, and genomic regions harboring 23 previously mapped QTL were narrowed down from an average of 4,052–1,844 kb.
Notably, Xie et al. (2010) developed a parent-independent genotyping method based on the principle of maximum parsimony of recombination (MPR). They sequenced a RIL population at a low coverage (0.055×), identified SNPs in RILs, and inferred the parental sources of each allele at all SNP loci with MPR. The resulting genotypes were further refined with a resampling procedure followed by Bayesian inference analysis. In common with the method developed by Andolfatto et al. (2011), the genotypes of 238 RILs were determined with the HMM approach, and a bin map was subsequently constructed for QTL mapping. A QTL for grain width was mapped to a bin of about 206 Kb, and this QTL coincided with a cloned gene GW5 for grain width, lending credence to this approach. The map constructed in this study was further used to locate QTL for traits of agronomic importance (Yu et al. 2011) and determine the genetic composition of yield heterosis (Zhou et al. 2012).
A WGR-based QTL mapping approach offers a gene-level mapping resolution in crops with a complex genome
Whole-genome duplications have occurred in the lineages of plants and are particularly widespread in the phylogeny of flowering plants (Tang et al. 2010). As a result, most crop genomes are more complicated than model plants, and a considerable number of genes are present in multiple copies in their genomes (Schmutz et al. 2010). These paralogous sequences make it difficult to map some NGS sequences to unique positions on a reference genome, and allelic variations cannot be distinguished from differences between paralogous sequences. Repetitive sequences, which account for more than 85 % of some crop genomes such as maize (Schnable et al. 2009), also present technical difficulties for WGR-based genotyping. They create ambiguities in alignment, which, in turn, produce incorrect genotyping results (Xu et al. 2013).
The issues caused by genome duplications and repetitive sequences were recently solved by an enhanced SNP validation and a haplotype-based QTL mapping in soybean, a palaeopolyploid crop that experienced two genome duplications (Xu et al. 2013). Figure 1 shows the workflow of this study, in which a RIL population was sequenced at a low coverage (~0.19×), and two parents were sequenced at a high coverage (~13.5×). SNPs identified between the two parents were validated by their segregation in the RIL population, and only those identified in both data sets were considered as candidate SNPs, which were subjected to Chi-sqaure tests to eliminate those potentially affected by misalignment, a consequence of genome duplications and repetitive sequences. The SNP set was further refined using the MPR method, and the RIL population was genotyped with the HMM approach.

Workflow of a biparental QTL mapping with the WGR approach in soybean. Two parental lines of a RIL population are sequenced at a high coverage and RILs at a low coverage. SNPs are identified between two parents, as well as RILs. Only those identified in both datasets are considered as candidate SNPs and subjected to Chi-square tests. SNPs segregating with a 1:1 ratio in the RIL population are further validated with a method based on the principle of maximum parsimony of recombination (MPR) and Bayesian inference. The phased SNPs are utilized to genotype the RIL population with a hidden Markov model (HMM). Based on genotyping results, recombination intervals were identified, and a bin map is subsequently constructed. Each bin harbors a set of SNPs and represents a haplotype, thus can be used to construct a linkage map for QTL mapping. All candidate SNPs are annotated, in term of synonymous and nonsynonymous SNPs. Once a QTL is mapped, the underlying genes can be putatively identified by the presence of nonsynonymous SNPs in genic regions and further pinpointed with gene expression experiments. QTNs or causal SNPs can be subsequently determined and used in marker-assisted selection
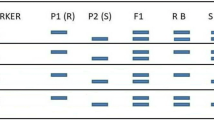
A crucial task of the genotyping procedure is to locate recombination intervals (RI), the genomic regions between two adjacent haplotype blocks (Fig. 2). In model crops having a compact genome, genotypic data in RIs were imputed with different algorithms (Andolfatto et al. 2011; Huang et al. 2009; Xie et al. 2010). In crops with a complex genome such as soybean, imputation is impractical and may compromise the power of QTL mapping because of the paucity of phased SNPs in repetitive regions, which results in larger RIs (Xu et al. 2013). Thus, instead of imputing data, a haplotype-based QTL mapping approach was proposed and utilized. Figure 2 shows the construction of a bin map. Based on genotyping results, RIs were delimited and bins were determined. Each bin harbors a number of SNPs and represents a haplotype, and thus can be used to construct the linkage map. The mapping of QTL for root-knot nematode resistance suggested that this haplotype-based approach provided a gene-level resolution, which was 16.7–144.5 times higher than the traditional method based on SNP array and SSR markers at three QTL loci, and the major QTL was mapped to a bin of 29.7 kb, which harbored three true genes and two pseudogenes. Based on SNP annotation, candidate genes and causal SNPs were pinpointed and confirmed by gene expression experiments.

Construction of a bin map. The green represents parent A allele, and the red represents parent B allele. The transition regions between two haplotype blocks (blank regions) are defined as recombination intervals (RI), and the genomic region between two adjacent RIs is defined as a bin. Each bin harbors a number of phased SNPs and represents a haplotype
The haplotype-based QTL mapping approach developed in soybean combines SNP discovery, SNP validation, and genotyping. Given that all sequence variations between two parental lines can be identified by deep sequencing and recombination events in each RIL can be subsequently determined, the mapping resolution is mainly dependent on population size. In soybean, when the RIL number increased from 246 to 499, the percentage of bins containing one to 10 genes increased from 50.8 to 81 % (Xu et al. 2013; Xu et al. unpublished data). Once a QTL is mapped to such a bin, there is a good chance to pinpoint the candidate genes underlying the QTL and identify causal SNPs, or QTNs, which can be readily employed in molecular breeding.
WGR-enabled genome-wide association studies
Genome-wide association (GWA) study has been widely used to identify loci for different diseases in human and has not been utilized in crops until recently. A major advantage of GWA is that it utilizes historical recombination events accumulated in the evolution of a set of diverse germplasm used in a study so that the mapping resolution may be significantly improved (Myles et al. 2009). GWA studies require genome-wide genotypes and phenotypes. The advent of effective genotyping platforms allowed the construction of HapMaps and made GWA a powerful QTL mapping approach in plants.
The early plant HapMaps were mainly constructed by expensive microarray-based approaches that suffered from substantial ascertainment biases (Clark et al. 2007; McNally et al. 2009; Kim et al. 2007). Recently, sequencing-based genotyping platforms were used to construct comprehensive HapMaps in rice (Huang et al. 2010, 2012a, b) and maize (Jiao et al. 2012; Chia et al. 2012) to facilitate GWA studies. Huang et al. (2010) first sequenced a set of 517 rice accessions at approximately 1× coverage using the Illumina sequencing platform. With an algorithm based on rice linkage disequilibrium (LD), they were able to impute missing data with a prediction accuracy of >98 % and subsequently constructed a rice HapMap. Based on this map, they performed GWA analysis and identified 80 loci for 14 agronomic traits, such as flower time and tiller. This study was extended by sequencing an additional 433 worldwide germplasm and examining genic sequence variations across the whole set of 950 accessions (Huang et al. 2012a). With an increased population size, the mapping power of GWA was improved, and 32 additional loci for 11 traits were identified. The low coverage sequences were further de novo assembled for functional annotation, which resulted in the identification of candidate genes for 18 loci, indicating that the WGR-based GWA has the potential to identify causal polymorphism. More recently, Huang et al. (2012b) further sequenced another 133 cultivated rice and 446 wild rice (Oryza rufipogon) accessions and identified 55 domestication sweeps and putative causal genes by analyzing all combined sequence data. In-depth analysis of these selection sweeps allowed them to determine the origins of cultivated rice, Oryza indica and Oryza japonica. They concluded that the O. japonica rice was first domesticated in the middle area of the Pearl River in southern China, and O. indica was subsequently developed from crosses between O. japonica and local wild rice as the initial cultivars that spread to Southeast Asia and South Asia. The WGR-based GWA was also highlighted in a study aimed at identifying QTL for metabolites in rice. Chen et al. (2014) performed comprehensive profiling of 840 metabolites in 529 diverse rice accessions and conducted GWA analysis using resequencing data. Among ~6.4 million SNP markers analyzed, they identified hundreds of sequence variants associated with numerous secondary metabolites with large effects and subsequently pinpointed 36 candidate genes that modulate levels of metabolites with potential physiological and nutritional importance.
GWA also offers the potential to rapidly resolve complex traits to gene-level resolution in maize, a major crop species of high genetic diversity (Gore et al. 2009). However, maize has a rapid LD decay (<2,000 bp), and thus, a high density of genome-wide markers is required for GWA studies. The first maize HapMap was constructed using sequence variations in low copy regions of 27 inbred lines that were utilized as founders of a nested association mapping population (Gore et al. 2009). The 1.6 million SNPs included in HapMap1 were employed to map QTL for flower time (Buckler et al. 2009), leaf architecture (Tian et al. 2011), stalk strength (Peiffer et al. 2013), plant height (Peiffer et al. 2014), southern leaf blight resistance (Kump et al. 2011), and northern leaf blight resistance (Poland et al. 2011). A large number of QTL with small additive effects were identified for each trait in these studies. For example, 30–36 QTL explaining >83 % of genetic variance were identified for leaf architecture traits, including leaf length, leaf width, and upper leaf angle. But causal variants for these QTL were rarely identified, suggesting that more SNPs are necessary to achieve better mapping resolution (Tian et al. 2011).
Recently, Jiao et al. (2012) sequenced 278 inbred lines at ~2× depth and obtained ~27.8 million SNPs. A subset of ~6.7 million SNPs with a missing data rate of <50 % were further analyzed, imputed, and utilized for a GWA study of cob color, silk color and date to anthesis. Some identified loci coincided with the expected targets known to influence these traits. Driven by GWA studies, the maize HapMap2 was constructed by sequencing 103 lines at ~4× depth (Chia et al. 2012). HapMap2 comprises 55 million SNPs and a large number of structure variations. Compared with HapMap1, sequence variants of HapMap2 are closer to saturating the genome with polymorphic markers in high LD, which is a prerequisite for GWA studies, and allows GWA to be performed in maize. HapMap2 has proved to be more powerful than HapMap1. Five traits that were previously mapped with HapMap1 markers (Tian et al. 2011; Kump et al. 2011; Poland et al. 2011) were reanalyzed with HapMap2 markers, and stronger trait–marker associations were observed, and two times more significantly associated loci were identified. Thus, it is expected that HapMap2 will greatly facilitate GWA studies in maize. Notably, structure variations are pervasive in maize genomes and account for 3.5 % of HapMap2 markers. These structure variations were enriched at loci associated with important traits and contributed 15–27 % of QTL for them (Chia et al. 2012), suggesting that identification of structure variations with the WGR approach is imperative for uncovering the genetic basis of important traits.
WGR allows rapid detection of causal mutations
Screening of a mutagenized population to identify causal sequence variants responsible for mutant phenotypes is an important method for gene discovery. Traditionally, this approach involves two discrete steps: mutation mapping and candidate gene sequencing. Recently, the WGR strategy has been widely used to identify causal mutations in model species (Schneeberger et al. 2009; Austin et al. 2011; Uchida et al. 2011). These approaches mainly combine NGS with BSA, a method for rapid identification of molecular markers associated with a gene of interest (Michelmore et al. 1991). The main task of these approaches is to deep sequence a DNA pool of F2 mutant plants derived from a cross between the mutant and a wild type. Given that all selected plants show mutant phenotypes, it is assumed that there is no or little recombination in the target genomic region of these plants. Thus, when aligning the sequence reads against the reference sequence to determine causal sequence variations of the pooled sample, tracts homozygous for the allele of the mutant (or enrichment of mutant allele) are indicative of no recombination events occurring in those regions, which should be within physical proximity of the mutations. Further examination of sequence variations between the pooled sample, and the reference sequence in the identified region will allow the identification of causal variants. SHOREmap, an extension of short-read sequence analysis pipeline SHORE, and the next-generation mapping (NGM) pipeline have been developed to identify causal mutations in A. thaliana (Schneeberger et al. 2009; Austin et al. 2011).
A similar approach, MutMap, was developed in rice (Abe et al. 2012; Fekih et al. 2013). In common with SHOREmap and NGM, this approach combines BSA and WGR to identify causal mutations. Abe and colleagues crossed a pale green leaf mutant with its progenitor wild type to derive an F2 mapping population. A pooled DNA sample from mutants was sequenced at high coverage, and causal SNPs were subsequently identified in OsCA01 gene for pale green leaf. The MutMap approach was also successfully used to identify a causal PAV for rice blast resistance gene Pii (Takagi et al. 2013b). In addition, this strategy was further extended to biparental population mapping in a method known as QTL-seq, which is based on WGR of two pooled DNA samples from the phenotypic extremes of a mapping population, either an F2 or RIL population (Takagi et al. 2013a). Using QTL-seq, Takagi et al. rapidly identified QTL for rice blast disease resistance and seedling vigor, which were further confirmed by classic QTL mapping. Recently, Liu et al. (2012) demonstrated how this strategy accelerated gene cloning in maize. Instead of genomic DNA sequence, they utilized RNA sequence to identify sequence variants at gene expression level between two bulks contrast in presence or absence of wax on maize juvenile leaves and successfully cloned the causal gene gl3.
The sequencing strategy was also applied to TILLING (Oleykowski et al. 1998), a functional genomics approach designed to discover rare mutations in rice (Till et al. 2007), wheat (Slade et al. 2005; Uauy et al. 2009), oat (Chawade et al. 2010), soybean (Cooper et al. 2008), sorghum (Xin et al. 2008), and Brassica (Stephenson et al. 2010). With a classic TILLING protocol, mutations are identified by the detection of PCR products digested at mismatched sites in heteroduplexes with CELI endonuclease (Oleykowski et al. 1998). Tsai et al. (2011) developed a method designated as TILLING by sequencing (Tsai et al. 2011), in which DNA samples of 768 mutagenized plants were multidimensionally pooled, and target genes were amplified and sequenced with a next-generation sequencer. This work led to the discovery of rare mutants in rice and wheat.
The combination of sequencing with BSA and TILLING greatly accelerates the discovery of mutation and causal sequence variations that are imperative for our understanding of genetic basis of phenotypic variation. We envision that this strategy will be widely used in more economically important crops in the near future and help develop germplasm of agronomic importance.
The perspective of using the third-generation sequencing technologies to realize the full potential of WGR approaches
NGS technologies have led the way in revolutionizing SNP discovery, molecular mapping, and gene discovery. Nevertheless, the inherent disadvantages of NGS technologies, such as short sequence reads and sequencing biases introduced by PCR amplifications, prevent realization of the full potential of WGR-based approaches in major crops, especially those with a complex genome. The third-generation sequencing (TGS) technologies have the potential to overcome these issues. Different from NGS technologies that rely on PCR to grow clusters of a given DNA template that are subsequently sequenced by synthesis, TGS technologies interrogate single molecules of DNA, such that sequencing biases caused by PCR can be avoided (Schadt et al. 2010). Currently available TGS platforms include Helicos true single-molecule sequencing (tsMS™) (Harris et al. 2008), Pacific BioSciences single-molecule real-time (SMRT™) sequencing (Eid et al. 2009), and Oxford Nanopores GridION and MinION systems (Clarke et al. 2009). Both Pacific BioSciences and Oxford Nanopores instruments can generate multiple kilobase sequence reads and have the potential to facilitate de novo sequencing, simplify sequence assembly by spanning repetitive sequence regions, detect CNVs, and improve the precision of WGR-based genotyping. Moreover, SMRT™ sequencing allows direct identification of nucleotide modification such as DNA methylation (Flusberg et al. 2010) and will greatly promote the application of epigenetics in plant breeding. Currently, the short sequence reads produced by NGS platforms are the major factor contributing to the low mapping resolution of the WGR-based genotyping approach in duplicated or repetitive genomic regions (Xu et al. 2013). However, the TGS technologies have not been widely utilized yet mainly because of higher sequencing error rates. To solve this issue, Koren and colleagues (2012) developed a new hybrid method that combines NGS and TGS technologies to produce long sequence reads with high accuracy. With continuous improvement of TGS technologies, we expect great enhancement in sequencing accuracy and output and significant reduction in sequencing costs. TGS technologies may help to realize the full potential of WGR-based approaches in sequence variation discovery and gene mapping and cloning in the future.
Conclusions
With the releases of many crop reference genome sequences, WGR has been extensively conducted to study genomic diversity and evolution. These studies have rapidly expanded our knowledge of genetic variations in crops and provided abundant genomic resources for genetic studies. The WGR-based QTL mapping approaches have turned the traditional three-step paradigm of marker discovery, marker validation, and genotyping into a single sequencing step and generated maps of a gene-level resolution in major crops, including those with a complex genome. WGR has also facilitated the construction of high-quality HapMaps in rice and maize, which allowed the performance of GWA studies and led to uncovering genetic variations underlying traits of agronomic importance. The combination of WGR with traditional BSA approach provides powerful tools for rapid identification of genes or causal mutations. In light of advances in TGS technologies, we envision that the WGR approaches will play a more pivotal role in QTL mapping and gene discovery and subsequently accelerate crop breeding.
Abbreviations
- BSA:
-
Bulked segregant analysis
- CNV:
-
Copy number variation
- GBS:
-
Genotyping by sequencing
- GWA:
-
Genome-wide association
- HMM:
-
Hidden Markov model
- LD:
-
Linkage disequilibrium
- MPR:
-
Maximum parsimony of recombination
- MSG:
-
Multiplexed shotgun genotyping
- NGM:
-
Next-generation mapping
- NGS:
-
Next-generation sequencing
- PAV:
-
Presence/absence variation
- QTL:
-
Quantitative trait loci
- QTN:
-
Quantitative trait nucleotides
- RIL:
-
Recombinant inbred line
- RRS:
-
Reduced representation sequencing
- SMRT™:
-
Single-molecule real-time sequencing
- SNP:
-
Single nucleotide polymorphism
- TGS:
-
Third-generation sequencing
- TILLING:
-
Targeting induced local lesions in genomes
- tsMS™:
-
True single-molecule sequencing
- WGR:
-
Whole-genome resequencing
References
Abe A, Kosugi S, Yoshida K, Natsume S, Takagi H, Kanzaki H et al (2012) Genome sequencing reveals agronomically important loci in rice using MutMap. Nat Biotechnol 30(2):174–178
Andolfatto P, Davison D, Erezyilmaz D, Hu TT, Mast J, Sunayama-Morita T, Stern DL (2011) Multiplexed shotgun genotyping for rapid and efficient genetic mapping. Genome Res 21(4):610–617
Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408(6814):796–815
Austin RS, Vidaurre D, Stamatiou G, Breit R, Provart NJ, Bonetta D et al (2011) Next-generation mapping of Arabidopsis genes. Plant J Cell Mol Biol 67(4):715–725
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA et al (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One 3(10):e3376
Bernardo R (2008) Molecular markers and selection for complex traits in plants: learning from the last 20 years. Crop Sci 48(5):1649
Buckler ES, Holland JB, Bradbury PJ, Acharya CB, Brown PJ, Browne C et al (2009) The genetic architecture of maize flowering time. Science 325(5941):714–718
Chawade A, Sikora P, Bräutigam M, Larsson M, Vivekanand V, Nakash MA et al (2010) Development and characterization of an oat TILLING-population and identification of mutations in lignin and beta-glucan biosynthesis genes. BMC Plant Biol 10(1):86
Chen W, Gao Y, Xie W, Gong L, Lu K, Wang W et al (2014) Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nat Genet 46(7):714–721
Chia J-M, Song C, Bradbury PJ, Costich D, de Leon N, Doebley J et al (2012) Maize HapMap2 identifies extant variation from a genome in flux. Nat Genet 44(7):803–807
Clark RM, Schweikert G, Toomajian C, Ossowski S, Zeller G, Shinn P et al (2007) Common sequence polymorphisms shaping genetic diversity in Arabidopsis thaliana. Science 317(5836):338–342
Clarke J, Wu H-C, Jayasinghe L, Patel A, Reid S, Bayley H (2009) Continuous base identification for single-molecule nanopore DNA sequencing. Nat Nanotechnol 4(4):265–270
Cook DE, Lee TG, Guo X, Melito S, Wang K, Bayless AM et al (2012) Copy number variation of multiple genes at Rhg1 mediates nematode resistance in soybean. Science 338(6111):1206–1209
Cooper JL, Till BJ, Laport RG, Darlow MC, Kleffner JM, Jamai A et al (2008) TILLING to detect induced mutations in soybean. BMC Plant Biol 8(1):9
Deschamps S, la Rota M, Ratashak JP, Biddle P, Thureen D, Farmer A et al (2010) Rapid genome-wide single nucleotide polymorphism discovery in soybean and rice via deep resequencing of reduced representation libraries with the Illumina genome analyzer. Plant Genome 3(1):53–68
Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G et al (2009) Real-time DNA sequencing from single polymerase molecules. Science 323(5910):133–138
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6(5):e19379. doi:10.1371/journal.pone.0019379
Fekih R, Takagi H, Tamiru M, Abe A, Natsume S, Yaegashi H et al (2013) MutMap+: genetic mapping and mutant identification without crossing in rice. PLoS One 8(7):e68529. doi:10.1371/journal.pone.0068529
Feltus FA, Wan J, Schulze SR, Estill JC, Jiang N, Paterson AH (2004) An SNP resource for rice genetics and breeding based on subspecies indica and japonica genome alignments. Genome Res 14(9):1812–1819
Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA et al (2010) Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods 7(6):461–465
Gao Z-Y, Zhao S-C, He W-M, Guo L-B, Peng Y-L, Wang J-J et al (2013) Dissecting yield-associated loci in super hybrid rice by resequencing recombinant inbred lines and improving parental genome sequences. Proc Natl Acad Sci USA 110(35):14492–14497
Goff SA, Ricke D, Lan T-H, Presting G, Wang R, Dunn M et al (2002) A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296(5565):92–100
Gore MA, Chia J-M, Elshire RJ, Sun Q, Ersoz ES, Hurwitz BL et al (2009) A first-generation haplotype map of maize. Science 326(5956):1115–1117
Harris TD, Buzby PR, Babcock H, Beer E, Bowers J, Braslavsky I et al (2008) Single-molecule DNA sequencing of a viral genome. Science 320(5872):106–109
Huang X, Feng Q, Qian Q, Zhao Q, Wang L, Wang A et al (2009) High-throughput genotyping by whole-genome resequencing. Genome Res 19(6):1068–1076
Huang X, Wei X, Sang T, Zhao Q, Feng Q, Zhao Y et al (2010) Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet 42(11):961–967
Huang X, Zhao Y, Wei X, Li C, Wang A, Zhao Q et al (2012a) Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat Genet 44(1):32–39. doi:10.1038/ng.1018
Huang X, Kurata N, Wei X, Wang Z-X, Wang A, Zhao Q et al (2012b) A map of rice genome variation reveals the origin of cultivated rice. Nature 490(7421):497–501
Hyten DL, Cannon SB, Song Q, Weeks N, Fickus EW, Shoemaker RC et al (2010) High-throughput SNP discovery through deep resequencing of a reduced representation library to anchor and orient scaffolds in the soybean whole genome sequence. BMC Genomics 11(1):38. doi:10.1186/1471-2164-11-38
International Rice Genome Sequencing Project (2005) The map-based sequence of the rice genome. Nature 436(7052):793–800
Jiao Y, Zhao H, Ren L, Song W, Zeng B, Guo J et al (2012) Genome-wide genetic changes during modern breeding of maize. Nat Genet 44(7):812–815
Kim S, Plagnol V, Hu TT, Toomajian C, Clark RM, Ossowski S et al (2007) Recombination and linkage disequilibrium in Arabidopsis thaliana. Nat Genet 39(9):1151–1155
Koren S, Schatz MC, Walenz BP, Martin J, Howard JT, Ganapathy G et al (2012) Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat Biotechnol 30(7):693–700
Kump KL, Bradbury PJ, Wisser RJ, Buckler ES, Belcher AR, Oropeza-Rosas MA et al (2011) Genome-wide association study of quantitative resistance to southern leaf blight in the maize nested association mapping population. Nat Genet 43(2):163–168
Lai J, Li R, Xu X, Jin W, Xu M, Zhao H et al (2010) Genome-wide patterns of genetic variation among elite maize inbred lines. Nat Genet 42(11):1027–1030
Lam H-M, Xu X, Liu X, Chen W, Yang G, Wong F-L et al (2010) Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat Genet 42(12):1053–1059
Lander ES, Botstein D (1989) Mapping mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121(1):185–199
Li F, Fan G, Wang K, Sun F, Yuan Y, Song G et al (2014) Genome sequence of the cultivated cotton Gossypium arboreum. Nat Genet 46(6):567–572. doi:10.1038/ng.2987
Liu S, Yeh C-T, Tang HM, Nettleton D, Schnable PS (2012) Gene mapping via bulked segregant RNA-Seq (BSR-Seq). PLoS One 7(5):e36406
Mayer KFX, Waugh R, Brown JWS, Schulman A, Langridge P, Platzer M et al (2012) A physical, genetic and functional sequence assembly of the barley genome. Nature 491(7426):711–716. doi:10.1038/nature11543
McNally KL, Childs KL, Bohnert R, Davidson RM, Zhao K, Ulat VJ et al (2009) Genomewide SNP variation reveals relationships among landraces and modern varieties of rice. Proc Natl Acad Sci USA 106(30):12273–12278
Michelmore RW, Paran I, Kesseli RV (1991) Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proc Natl Acad Sci 88(21):9828–9832
Myles S, Peiffer J, Brown PJ, Ersoz ES, Zhang Z, Costich DE, Buckler ES (2009) Association mapping: critical considerations shift from genotyping to experimental design. Plant Cell 21(8):2194–2202
Oleykowski CA, Bronson Mullins CR, Godwin AK, Yeung AT (1998) Mutation detection using a novel plant endonuclease. Nucleic Acids Res 26(20):4597–4602
Peiffer JA, Flint-Garcia SA, De Leon N, McMullen MD, Kaeppler SM, Buckler ES (2013) The genetic architecture of maize stalk strength. PLoS One 8(6):e67066. doi:10.1371/journal.pone.0067066
Peiffer JA, Romay MC, Gore MA, Flint-Garcia SA, Zhang Z, Millard MJ et al (2014) The genetic architecture of maize height. Genetics 196(4):1337–1356. doi:10.1534/genetics.113.159152
Poland JA, Rife TW (2012) Genotyping-by-sequencing for plant breeding and genetics. Plant Genome J 5(3):92. doi:10.3835/plantgenome2012.05.0005
Poland JA, Bradbury PJ, Buckler ES, Nelson RJ (2011) Genome-wide nested association mapping of quantitative resistance to northern leaf blight in maize. Proc Natl Acad Sci USA 108(17):6893–6898
Poland JA, Brown PJ, Sorrells ME, Jannink J-L (2012a) Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS One 7(2):e32253. doi:10.1371/journal.pone.0032253
Poland J, Endelman J, Dawson J, Rutkoski J, Wu S, Manes Y et al (2012b) Genomic selection in wheat breeding using genotyping-by-sequencing. Plant Genome J 5(3):103. doi:10.3835/plantgenome2012.06.0006
Potato Genome Sequencing Consortium (2011) Genome sequence and analysis of the tuber crop potato. Nature 475(7355):189–195
Schadt EE, Turner S, Kasarskis A (2010) A window into third-generation sequencing. Hum Mol Genet 19(R2):R227–R240
Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W et al (2010) Genome sequence of the palaeopolyploid soybean. Nature 463(7278):178–183
Schmutz J, McClean PE, Mamidi S, Wu GA, Cannon SB, Grimwood J et al (2014) A reference genome for common bean and genome-wide analysis of dual domestications. Nat Genet 46(7):707–713. doi:10.1038/ng.3008
Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S et al (2009) The B73 maize genome: complexity, diversity, and dynamics. Science 326(5956):1112–1115
Schneeberger K, Ossowski S, Lanz C, Juul T, Petersen AH, Nielsen KL et al (2009) SHOREmap: simultaneous mapping and mutation identification by deep sequencing. Nat Methods 6(8):550–551
Slade AJ, Fuerstenberg SI, Loeffler D, Steine MN, Facciotti D (2005) A reverse genetic, nontransgenic approach to wheat crop improvement by TILLING. Nat Biotechnol 23(1):75–81
Stephenson P, Baker D, Girin T, Perez A, Amoah S, King GJ, Østergaard L (2010) A rich TILLING resource for studying gene function in Brassica rapa. BMC Plant Biol 10(1):62. doi:10.1186/1471-2229-10-62
Takagi H, Abe A, Yoshida K, Kosugi S, Natsume S, Mitsuoka C et al (2013a) QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J 74(1):174–183
Takagi H, Uemura A, Yaegashi H, Tamiru M, Abe A, Mitsuoka C et al (2013b) MutMap-Gap: whole-genome resequencing of mutant F2 progeny bulk combined with de novo assembly of gap regions identifies the rice blast resistance gene Pii. New Phytol 200(1):276–283
Tang H, Bowers JE, Wang X, Paterson AH (2010) Angiosperm genome comparisons reveal early polyploidy in the monocot lineage. Proc Natl Acad Sci USA 107(1):472–477. doi:10.1073/pnas.0908007107
Tian F, Bradbury PJ, Brown PJ, Hung H, Sun Q, Flint-Garcia S et al (2011) Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat Genet 43(2):159–162
Till BJ, Cooper J, Tai TH, Colowit P, Greene EA, Henikoff S, Comai L (2007) Discovery of chemically induced mutations in rice by TILLING. BMC Plant Biol 7(1):19
Tomato Genome Consortium (2012) The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485(7400):635–641. doi:10.1038/nature11119
Tsai H, Howell T, Nitcher R, Missirian V, Watson B, Ngo KJ et al (2011) Discovery of rare mutations in populations: TILLING by sequencing. Plant Physiol 156:1257–1268
Uauy C, Paraiso F, Colasuonno P, Tran RK, Tsai H, Berardi S et al (2009) A modified TILLING approach to detect induced mutations in tetraploid and hexaploid wheat. BMC Plant Biol 9(1):115
Uchida N, Sakamoto T, Kurata T, Tasaka M (2011) Identification of EMS-induced causal mutations in a non-reference Arabidopsis thaliana accession by whole genome sequencing. Plant Cell Physiol 52(4):716–722
Van Orsouw NJ, Hogers RCJ, Janssen A, Yalcin F, Snoeijers S, Verstege E et al (2007) Complexity reduction of polymorphic sequences (CRoPS): a novel approach for large-scale polymorphism discovery in complex genomes. PLoS One 2(11):e1172. doi:10.1371/journal.pone.0001172
Van Tassell CP, Smith TPL, Matukumalli LK, Taylor JF, Schnabel RD, Lawley CT et al (2008) SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries. Nat Methods 5(3):247–252
Varshney RK, Chen W, Li Y, Bharti AK, Saxena RK, Schlueter JA et al (2012) Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat Biotechnol 30(1):83–89
Varshney RK, Song C, Saxena RK, Azam S, Yu S, Sharpe AG et al (2013) Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat Biotechnol 31(3):240–246
Xie W, Feng Q, Yu H, Huang X, Zhao Q, Xing Y et al (2010) Parent-independent genotyping for constructing an ultrahigh-density linkage map based on population sequencing. Proc Natl Acad Sci USA 107(23):10578–10583
Xin Z, Wang ML, Barkley NA, Burow G, Franks C, Pederson G, Burke J (2008) Applying genotyping (TILLING) and phenotyping analyses to elucidate gene function in a chemically induced sorghum mutant population. BMC Plant Biol 8(1):103. doi:10.1186/1471-2229-8-103
Xu X, Liu X, Ge S, Jensen JD, Hu F, Li X et al (2012) Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat Biotechnol 30(1):105–111
Xu X, Zeng L, Tao Y, Vuong T, Wan J, Boerma R et al (2013) Pinpointing genes underlying the quantitative trait loci for root-knot nematode resistance in palaeopolyploid soybean by whole genome resequencing. Proc Natl Acad Sci USA 110(33):13469–13474
Yu J, Hu S, Wang J, Wong GK-S, Li S, Liu B et al (2002) A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 296(5565):79–92
Yu H, Xie W, Wang J, Xing Y, Xu C, Li X et al (2011) Gains in QTL detection using an ultra-high density SNP map based on population sequencing relative to traditional RFLP/SSR markers. PLoS One 6(3):e17595. doi:10.1371/journal.pone.0017595
Zhou G, Chen Y, Yao W, Zhang C, Xie W, Hua J et al (2012) Genetic composition of yield heterosis in an elite rice hybrid. Proc Natl Acad Sci USA 109(39):15847–15852
Acknowledgments
Mentioning of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the US Department of Agriculture. USDA is a equal opportunity provider and employer.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Xu, X., Bai, G. Whole-genome resequencing: changing the paradigms of SNP detection, molecular mapping and gene discovery. Mol Breeding 35, 33 (2015). https://doi.org/10.1007/s11032-015-0240-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-015-0240-6