
Abstract
Ascochyta blight, caused by the fungus Ascochyta rabiei (Pass.) Labr., is a highly destructive disease of chickpea (Cicer arietinum L.) on a global basis, and exhibits considerable natural variation for pathogenicity. Different sources of ascochyta blight resistance are available within the cultivated species, suitable for pyramiding to improve field performance. Robust and closely linked genetic markers are desirable to facilitate this approach. A total of 4,654 simple sequence repeat (SSR) and 1,430 single nucleotide polymorphism (SNP) markers were identified from a chickpea expressed sequence tag (EST) database. Subsets of 143 EST–SSRs and 768 SNPs were further used for validation and subsequent high-density genetic mapping of two intraspecific mapping populations (Lasseter × ICC3996 and S95362 × Howzat). Comparison of the linkage maps to the genome of Medicago truncatula revealed a high degree of conserved macrosynteny. Based on field evaluation of ascochyta blight incidence performed over 2 years, two genomic regions containing resistance determinants were identified in the Lasseter × ICC3996 family. In the S95362 × Howzat population, only one quantitative trait locus (QTL) region was identified for both phenotypic evaluation trials, which on the basis of bridging markers was deduced to coincide with one of the Lasseter × ICC3996 QTLs. Of the two QTL-containing regions identified in this study, one (ab_QTL1) was predicted to be in common with QTLs identified in prior studies, while the other (ab_QTL2) may be novel. Markers in close linkage to ascochyta blight resistance genes that have been identified in this study can be further validated and effectively implemented in chickpea breeding programs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cultivated chickpea (Cicer arietinum L.) is a highly valuable global food crop that provides a good source of protein, dietary fibre, carbohydrates and minerals. Chickpea is a self-pollinating annual plant species capable, like other legumes, of improving soil fertility through fixation of atmospheric nitrogen. As a crop of semi-arid regions, it can be grown on non-irrigated farmland, providing an effective source of dietary protein for humans, while stubble residues may also be used for animal fodder. Chickpea seed can be highly diverse in morphological terms. However, only two major types (morphotypes) are cultivated, termed Desi and Kabuli. Desi kernels are smaller and darker in colour, while Kabuli kernels are larger and rounder, and they are usually beige or cream in colour (Moreno and Cubero 1978; Sefera et al. 2011). In Australia, chickpea is cultivated in the eastern and southern regions of the continent, and is used as a rotation crop by many farmers. To date, both Australian-based and global chickpea production industries have been hindered by several damaging diseases including phytophthora root rot, viruses, botrytis grey mould and ascochyta blight, the last being particularly devastating. Different international chickpea breeding programs have obtained superior varieties which exhibit enhanced resistance to some diseases, but conventional breeding for such outcomes is a time-consuming and laborious exercise. Genomics-assisted breeding strategies are therefore required to develop superior varieties with greater disease resistance in a reduced time frame.
Compared to other cool-season legume crops such as lentil, field pea and faba bean, genomic resources supporting effective molecular genetic marker-based breeding and identification of disease resistance genes for chickpea are at present relatively abundant. To date, several transcriptome sequencing studies for cultivated and non-domesticated chickpea varieties have been completed (Hiremath et al. 2011; Garg et al. 2011; Jhanwar et al. 2012) and reference genome sequences have recently been determined (Jain et al. 2013; Varshney et al. 2013). Although valuable, such information still requires subsequent marker development and integration into breeding programs for deployment in crop improvement programs.
Genetic map construction for use in marker-assisted breeding has been a part of breeding programs for major crops over several decades. So far, there has been a large emphasis on maps based on interspecific crosses, particularly those between cultivated chickpea C. arietinum and a non-domesticated species, C. reticulatum L. A saturated reference genetic linkage map of this type has been constructed through the use of simple sequence repeat (SSR) and single nucleotide polymorphism (SNP) markers (Thudi et al. 2011; Gaur et al. 2011, 2012). However, despite significant efforts in marker development, few high-density intraspecific linkage maps are available, in comparison to the status of many other legume species such as soybean (Glycine max [L.] Merr.), cowpea (Vigna unguiculata [L.] Walp) and common bean (Phaseolus vulgaris L.) (Thudi et al. 2011). The rationale for the use of interspecific mapping populations has been the low levels of genetic diversity that are known to be present within the gene pool of cultivated chickpea (Labdi et al. 1996; Thudi et al. 2011; Jhanwar et al. 2012). However, interspecific mapping populations are liable to detection of quantitative trait loci (QTLs) of major effect on domestication-related traits, which are unlikely to be present in cultivated germplasm, limiting the probability of successful information transfer into breeding programs (Cong et al. 2002; van der Knaap and Tanksley 2003). For chickpea, therefore, an intraspecific genetic linkage map constructed from a cross between genotypes from the cultivated gene pool would be more valuable for breeding applications.
To date, no chickpea intraspecific reference linkage map based on gene-based markers such as SNPs has been constructed, although some recently published interspecific linkage maps have been populated with such markers (Thudi et al. 2011; Choudhary et al. 2012a). Until recently, SSRs have been the marker system of choice because of their abundance, reproducibility and co-dominant nature, but recently advances in sequencing technology and multiplexing capabilities have increased the popularity of SNP markers for construction of genetic maps and genetic diversity studies. SNP markers offer many advantages, being generally biallelic in nature, co-dominantly inherited and present at high density within genomes. In addition, discovery of SNP loci from transcribed regions of the genome can provide a direct link between sequence polymorphism and putative functional variation. High-density genetic maps constructed from gene-based markers hence represent a powerful resource for genome analysis, providing an important opportunity to directly identify genes related to agronomic traits (Choudhary et al. 2012b). However, only a limited number of reports are available for expressed sequence tag (EST)-derived markers of chickpea (Buhariwalla et al. 2005; Choudhary et al. 2009; Varshney et al. 2009; Nayak et al. 2010; Gujaria et al. 2011). For this reason, enrichment of transcribed sequence resources to generate EST-based functional markers will be important to increase both marker resources and marker density for detection of important genomic loci in chickpea.
Ascochyta blight, caused by the fungus Ascochyta rabiei (Pass.) Labr. [teleomorph: Didymella rabiei (Kovacheski) von Arx (synonym: Mycosphaerella rabiei Kovacheski)] is managed through crop rotation, hygiene, seed treatment, preventive fungicide application and planting of varieties with improved resistance. Various molecular marker-based studies have identified QTLs for ascochyta blight resistance through intraspecific genetic mapping (Udupa and Baum 2003; Iruela et al. 2007; Anbessa et al. 2009; Madrid et al. 2012). However, to date, only a limited number of gene-based markers capable of efficient implementation within a breeding program have been described in close linkage to ascochyta blight resistance determinants segregating within cultivated germplasm (Madrid et al. 2013).
The current study describes the following steps: transcriptome sequencing of four distinct chickpea genotypes, followed by development of both SSR and SNP markers; construction of genetic linkage maps from two chickpea mapping populations (Lasseter × ICC3996, S95362 × Howzat) that segregate for ascochyta blight resistance; determination of conserved synteny with the M. truncatula genome; and identification of QTLs associated with ascochyta blight resistance, along with linked genetic loci suitable for marker-assisted selection. The implications for effective use in chickpea germplasm enhancement programs are discussed.
Materials and methods
Plant material and DNA extraction
Four chickpea genotypes (Lasseter, ICC3996, S95362, Howzat) exhibiting various differences in levels of resistance to ascochyta blight were selected to perform transcriptome sequencing, linkage mapping and QTL analysis. Two F6 recombinant inbred line (RIL) populations were generated from intraspecific crosses between chickpea genotypes, followed by single seed descent. The Lasseter (ascochyta blight-susceptible Desi type) × ICC3996 (ascochyta blight-resistant Desi type) and S95362 (ascochyta blight-resistant Kabuli type) × Howzat (ascochyta blight- moderately susceptible Desi type) populations contained 150 and 119 individuals, respectively. Leaf material was harvested from young plants, and genomic DNA was extracted using the Qiagen DNeasy 96 Plant Kit according to the manufacturer’s instructions. DNA was eluted into 80 μl of sterile water and stored at −20 °C until required.
Transcriptome sequencing and marker discovery
Transcriptome sequencing
Each of the mapping parents (Lasseter, ICC3996, S95362, Howzat) was used to perform transcriptome sequencing. Different plant tissues were used for RNA isolation from chickpea plants at various developmental stages: seedlings, leaf (young and mature), stem, flowers, immature pods, mature pods and immature seeds. RNA isolation and cDNA library preparation was performed using protocols as described in Kaur et al. (2012). EST sequencing was performed using 454 GS-FLX Titanium technology following the manufacturer’s instructions (Roche Diagnostics). Sequence reads were assembled de novo using NextGene software (Softgenetics, State College, PA, USA), with adaptor and primer sequences being removed prior to assembly using the ‘trimming’ function (by trimming sequences with 100 % similarity to the primer/adaptor sequence). De novo assembly was performed using the Greedy algorithm and error correction condensation.
SSR discovery and validation
EST contigs generated as a result of de novo assembly of 454 transcriptome data were used for SSR detection. Primer pair design was performed using the methods and parameters described in Kaur et al. (2011, 2012). All forward primers were designed with M13 sequence to enable fluorescent labelling of the PCR products (Schuelke 2000).
For validation, a total of 96 SSRs were randomly selected and tested on five chickpea genotypes (four mapping parents and one genotype of a related non-domesticated species [Cicer reticulatum]). PCR reactions were performed as mentioned in Kaur et al. (2011, 2012). All PCR reactions were performed in a 384-well format using a Biomek FX liquid handling robot (Beckman Coulter, Inc., Brea, CA, USA) using custom protocols and were amplified using a Bio-Rad MJ PTC-200 thermocycler. Post-PCR pooling was conducted as appropriate, and all samples were run using an ABI 3730xl with the ABI GeneScan LIZ500 size standard (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s instructions. PCR product sizes for genotyping were determined using the GeneMapper® v3.7 software (Applied Biosystems).
SNP discovery and validation
SNP prediction was performed from EST-contigs using NextGENe software v1.96 (Softgenetics). Quality-trimmed reads from all four cultivars were reference-aligned against the EST contig database using the ‘Alignment’ tool and ‘SNP/Indel discovery’ applications to detect all possible base variants, which were further filtered to obtain a subset of high quality SNPs. Any base variants that segregated within a genotype and all insertion–deletion mutants (indels) were excluded. The data set was further filtered on the basis of sequencing depth (>6 reads) and presence of other sequence variants within 20 bp flanking the targeted SNP, in order to obtain a set of high-confidence SNPs.
A subset of 48 SNPs was validated for all mapping parents using a direct Sanger sequencing approach. Forward and reverse primer pairs were designed corresponding to the sequences flanking the target SNPs using Primer3 sofware and Sequencher v4.10.1 (Gene Codes Corporation, MI, USA). PCR amplification was performed in a 12.5-μl reaction containing 20 ng DNA, 1× PCR buffer (Bioline), 15 pmol of each primer, 0.2 mM of each dNTP and 0.1 U IMMOLASE™ (Bioline). PCR conditions included a hot start at 95 °C for 10 min, followed by 35 cycles of 94 °C for 30 s, 46–50 °C for 30 s and 72 °C for 30 s, and a final elongation step of 72 °C for 10 min. PCR products were purified using 2.5 μl of shrimp alkaline phosphatase (10× SAP) and 0.1 μl of Exonuclease I (at 20 U/μl) per reaction. Purified PCR products were analysed using a sequencing primer and BigDye® Terminator v3.1 sequencing chemistry following the manufacturer’s instructions. Final PCR products were purified using ethanol precipitation, and resuspended in 12 μl Hi–Di formamide for sequence determination using an ABI3730xl (Applied Biosystems) capillary electrophoresis platform according to the manufacturer’s instructions. Sequence analysis and assembly of the resulting electropherograms was performed in Sequencher v4.10.1, and SNP validation was visually confirmed.
SSR and SNP genotyping
A total of 384 SSR marker assays, including publicly available and in-house developed SSRs (Supplementaries 1 and 2), were screened for polymorphism detection using PCR conditions and genotyping methods as described above.
Regions extending 100 bp 5′ and 3′ from the target sequence variant were selected for 823 putative SNP loci and submitted to Illumina for assay design. Of these, a total of 768 SNP loci were further selected for SNP genotyping based on design rank and score (Supplementary 3). Both mapping populations were SNP genotyped using the OPA tool in accordance with the manufacturer’s instructions. Custom genotyping assays were processed by the Illumina iScan reader, and analysis of SNPs and assignment of genotypes was carried out using GenomeStudio software v2011.1 (Illumina).
Genetic linkage mapping and linkage group nomenclature
All genotypic marker data was tested for conformation with the expected Mendelian segregation ratio of 1:1 using a Chi squared (χ2) test (P < 0.05). Linkage analysis was performed using Map Manager software version QTXb19 (Manly et al. 2001). Linkage groups (LGs) were constructed at a recombination fraction (q) of 0.25 and LOD score of 4.0. The order of markers on each LG was verified and confirmed using the “ripple” command. The Kosambi mapping function (Kosambi 1944) was used to convert the recombination fractions into additive genetic distance (centiMorgans). Mapchart (v2.1) was used for visualisation (Voorips 2002).
LGs were assigned, when possible, by extrapolation of known marker positions from reference linkage maps using publicly available markers as anchors (Gaur et al. 2011, 2012; Thudi et al. 2011). Each LG was also compared to the Medicago truncatula (Mt) genome, and the order of markers on each LG was further confirmed through comparative genomics analysis based on Mt chromosomal coordinates.
Evaluation of ascochyta blight resistance
The RIL populations and parents were sown in Horsham, Victoria, Australia in 2005 and 2009 in single rows of 5 m length in a randomised complete block design with three replicates. After establishment, plants were inoculated with ascochyta blight-infected stubble (mulched into 5-cm pieces) from the previous season. To ensure a uniform source of inoculum, a susceptible spreader row (of cv. Howzat) was sown every sixth row and the area surrounding the experiment was also sown with this variety. Disease symptoms were scored twice in 2005 and three times in 2009 using a scale of 1–9 based on whole plant severity (Singh et al. 1981). The final assessment score in each year was used to obtain the phenotypic assessment. Phenotypic assessment data was analysed to estimate means after adjustment for any spatial patterning within the trial. Models were fitted using residual maximum likelihood (REML) as implemented in GenStat (GenStat Committee, 2002 and previous releases). Means of symptom rating from each individual of the mapping populations were used to construct distribution histograms in order to determine the mode of inheritance for the trait.
QTL analysis and candidate gene selection
QTL detection was performed using marker regression, simple interval mapping (SIM) and composite interval mapping (CIM) in QTL Cartographer v2.5 (Wang et al. 2012). For SIM, an arbitrary LOD threshold of 2.5 was used to determine significance, while for CIM, significance levels for LOD thresholds were determined using 1,000 permutations.
The sequences that underpin the SNP loci flanking QTL intervals were BLAST analysed against the reference genome sequence of chickpea (Varshney et al. 2013) to identify candidate genes within the target region. The corresponding sequences were extracted from the chickpea reference genome and gene predictions were performed using the FGENESH program (www.Softberry.com). Predicted gene sequences were then BLAST analysed in NCBI to obtain functional annotations.
Results
Transcriptome sequencing, marker discovery and validation
A total of 1.43 × 106 reads were generated as a result of transcriptome sequencing from the four chickpea genotypes. After trimming of adaptor/primer sequences, as well as an additional 30–40 nucleotides from both the 5′ and 3′ termini of each sequence, a total of 1,261,642 high-quality reads were obtained for subsequent assembly. After clustering and assembly, a total of 20,880 contigs and 131,450 singletons were obtained. The unigene set was then further assessed for quality based on read length, and any remnant sequences less than 100 bp were excluded from further analysis, leaving a total of 92,164 unigenes comprised of 20,846 contigs and 71,596 singletons (Supplementaries 4 and 5). The length of the contigs ranged from 100 bp to 4,673 bp, with an average of 791 bp (Fig. 1) while the singleton read length varied from 100 to 548 bp, with an average of 269 bp (Fig. 2). Average contig coverage was 15-fold (ranging from 1.16- to 5787.14-fold) and the number of reads per contig varied between 2 and 51,569, with an average of 49.9 (Fig. 3).

Frequency histogram depicting the distribution of number of contigs as a function of read length

Frequency histogram depicting the distribution of number of singletons as a function of read length

Frequency histogram depicting the distribution of number of contigs as a function of number of reads
A total of 4,654 SSR primer pairs were obtained from in silico design, using EST-contigs as templates. A subset of 96 EST–SSR primer pairs was selected for validation of marker assay performance. A total of 78 primer pairs successfully obtained amplification products, of which 16 (20.5 %) revealed polymorphisms between the four C. arietinum genotypes. Inclusion of C. reticulatum, as the non-domesticated species, permitted polymorphism detection by 19 additional primer pairs (an increase to 44.8 % of the total; Supplementary 6).
A total of 25,673 base variants were predicted from comparison of transcriptome reads obtained from four mapping parents against the reference EST-containing contig database, and a SNP frequency of 1.55 SNPs per kb (at an average of 0.775 SNPs per kb between two haplotypes) was observed. After further filtering, a subset of 1,744 high-quality SNPs was obtained and of them 823 high-confidence SNPs were further selected for SNP-OPA design based on incidence of polymorphism between mapping family parents, and suitability for common use between the two populations. A final collection of 768 SNP loci was assembled that represented the optimal marker set, in terms of predicted assay performance, predicted genome coverage and polymorphism across the two maps under construction. Prior to synthesis of the 768-plex SNP-OPA, a subset of 48 SNP loci was evaluated through Sanger sequencing, of which 36 (75 %) exhibited successful amplicon production, and 28 (78 %) were validated in mapping parents (Fig. 4).

Example of SNP validation using Sanger sequencing approach showing the occurrence of two SNPs between different mapping parents
Genetic linkage mapping
The intraspecific mapping populations used for genotyping consisted of 150 and 119 individuals from Lasseter × ICC3996 and S95362 × Howzat, respectively. A total of 384 SSR marker assays, including 241 that were previously published (Table 1; Supplementary 1) and 143 EST-SSRs that were developed in this study, were screened for polymorphism detection (Supplementaries 1 and 2). A total of 14.84 % (Lasseter × ICC3996) and 13.02 % (S95362 × Howzat) of the SSR markers detected polymorphisms (Table 1). The rate of polymorphism was higher for publicly available SSRs (19 % for Lasseter × ICC3996; 17 % for S95362 × Howzat) than for EST-SSRs developed as part of this study (9 % for Lasseter × ICC3996; 6 % for S95362 × Howzat). Primer pairs revealing variation were then screened on the full progeny sets. A total of 57 and 50 SSRs were found to be polymorphic in the Lasseter × ICC3996 and S95362 × Howzat populations, respectively.
A common subset of 768 SNPs was screened on both mapping populations, of which 447 and 392 were polymorphic for Lasseter × ICC3996 and S95362 × Howzat, respectively (Table 1). Only 126 SNPs were found to be common between the two mapping populations. For each SNP, three main clusters were identified, corresponding to AA homozygotes, AB heterozygotes and BB homozygotes. The majority of the SNP markers produced two major clusters representing the two homozygous genotypes, with an occasional extra cluster corresponding to the heterozygous class (Supplementary 7). As both mapping populations were descended to the F6 level, the frequency of heterozygous combinations was expected to be low, as was observed in practice (c. 5 % in both populations).
In total, the SNP and SSR assays generated 504 (Lasseter × ICC3996) and 442 (S95362 × Howzat) markers suitable for genetic mapping analysis (Table 1). The Chi squared test (P < 0.05) identified 1.8 and 7.1 % markers from the Lasseter × ICC3996 and S95362 × Howzat populations, respectively, that did not segregate in accordance with the expected Mendelian inheritance ratio. All markers exhibiting such significant segregation distortion were excluded from the final analysis. The proportion of markers that were assigned to loci on LGs was 98.7 and 93 % for the Lasseter × ICC3996 and S95362 × Howzat maps, respectively. The remaining markers were unlinked. In total, nine LGs and three satellites were generated for Lasseter × ICC3996, and seven LGs and three satellites were obtained for S95362 × Howzat (Supplementaries 8 and 9). As high levels of colinearity were observed with previously published maps, most of the satellite LGs could be reasonably expected to be coalesced with the corresponding intact LG through the use of a larger number of markers (data not shown). The cumulative length of the Lasseter × ICC3996 map was 658.7 cM, with an average distance of 1.74 cM between loci, while the S95362 × Howzat map spanned a total length of 752 cM, with an average marker density of one locus per 2.16 cM (Table 2). All LGs from each map were compared for common marker loci (Supplementary 9). Publicly available SSRs were used as anchoring markers to identify individual LGs based on existing chickpea linkage map nomenclature.
Determination of conserved synteny with M. truncatula
All sequences underpinning the map-assigned genic SNP markers were compared to the genome draft of M. truncatula in order to characterise conserved macrosyntenic relationships. A total of 159 from 410 loci on the Lasseter × ICC3996 map and 131 from 363 loci on the S95362 × Howzat map were available for this analysis. A number of chickpea LGs exhibited macrosynteny to more than one Mt chromosome (Table 3). LG1 predominantly displayed blocks of synteny with MtChr2, with some additional affinities to MtChr3, 4 and 6 (Fig. 5). Moreover, LG2 contained substantial conservation of marker order with MtChr2, 4, 5 and 6 and LG3 displayed similar relationships with MtChr7. Most of the markers from LG4 showed matches to MtChr1, while LG5 was the syntenic counterpart to MtChr3. High levels of synteny were observed between LG6 and MtChr4, and LG7 showed a majority of matches to MtChr8. LG8 shared commonality with MtChr 5 and 6 (Supplementary 10).

Depiction of syntenic relationships between chickpea LGs and chromosomes of M. truncatula
Phenotypic analysis, QTL detection and candidate gene identification
Significant differences were observed in the necrosis rating (measured on a 1–9 scale) of whole plants among the RILs for each population at both times of evaluation intervals. A high degree of correlation (r 2 = 0.88 for Lasseter × ICC3996 and 0.75 for S95362 × Howzat) was observed in the data obtained from two time points of phenotypic assessment screens. Frequency distribution patterns obtained from both populations indicated the presence of multiple genes responsible for ascochyta blight resistance (Supplementary 11).
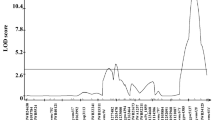
The locations and magnitudes of effect for each QTL were estimated using both SIM and CIM (Supplementary 12). On the Lasseter × ICC3996 map, CIM detected one QTL on LG4.1 for year 2005 and two QTLs on LGs 4.1 and 4.2 for year 2009 (Table 4), which in combination explained an estimated 45 and 26 % of phenotypic variance (V p) respectively. However, SIM analysis was able to detect a second QTL on LG4.2 from the 2005 data as well (Supplementary 12). On the S95362 × Howzat population-derived map, in contrast, a single QTL was detected on LG 4 through the use of CIM, accounting for totals of 59 % for year 2005 and 28 % for year 2009. This QTL appeared to coincide with the location of the QTL on LG4.1 from the Lasseter × ICC3996 map (Fig. 6; Table 4).

Localisation of QTLs on Lasseter × ICC3996 (ab2005/2009; LG4.1 and LG4.2) and S95362 × Howzat (ab2009; LG4) and identification of common marker loci between the two QTL-containing regions
All predicted QTLs were also compared to those from previously published studies of trait dissection for ascochyta blight resistance in chickpea. The QTL that was detected in common between Lasseter × ICC3996 (ab_2005/2009, LG4.1) and S95362 × Howzat (ab_2005/2009, LG4) was deduced to be located in an equivalent region to QTLs reported in earlier studies (Table 4).
BLAST analysis of sequences underpinning the SNP loci that flanked QTL intervals revealed matches to chromosome 4 of the chickpea reference genome sequence at distinct locations (Supplementary 13). Using gene prediction and BLAST analysis, a total of three candidate genes that display functional annotations as chitinase-like proteins were identified from the ab_QTL1-containing region of the Lasseter × ICC3996 map (Supplementary 14). Such proteins have been previously reported to be associated with quantitative variation in pathogen defence mechanisms in plants.
Discussion
Significance of genic SSR and SNP markers
In the areas of sequence polymorphism discovery and genetic map development, progress for chickpea has remained slow. Low levels of genetic diversity have been consistently described for the cultivated chickpea gene pool, and hence more specific marker resources are needed to enrich genetic maps in order to identify genes of interest (Labdi et al. 1996; Thudi et al. 2011; Jhanwar et al. 2012). Second-generation DNA sequencing technologies provide cost- and time-effective means for transcriptome sequencing and characterisation to further assist such marker discovery. The large majority of currently available EST-derived genic markers, including SSRs and SNPs, have only recently been developed from both cultivated and non-domesticated chickpea sources (Choudhary et al. 2012b; Hiremath et al. 2012; Jhanwar et al. 2012). The present study consequently provides a major source of additional genetic markers for chickpea molecular breeding.
The average contig length produced in this study was 791 bp, comparable to that from similar studies (523 bp: Garg et al. 2011; 459 bp: Hiremath et al. 2011; 946 bp: Jhanwar et al. 2012). All of the generated EST contigs were further used for the large-scale identification and validation of EST-derived SSRs and SNPs. When tested across a panel of four mapping family parents, the polymorphism rate for EST–SSRs was c. 21 %, comparable to the results of previous studies by Choudhary et al. (2009, 2012a), which reported 17 and 28 % EST-SSR polymorphism rates for intra- (C. arietinum) and interspecies (C. arietinum and C. reticulatum) comparison, respectively. In both populations the rate of polymorphism was higher for public SSRs that were of genomic origin, compared to EST–SSRs developed in the current study, which is in accordance with many other similar studies (Chabane et al. 2005, Mattioni et al. 2010). Although lower polymorphism rates were obtained for EST–SSRs, they were still sufficient to support genetic map construction. EST–SSRs exhibit many advantages over genomic SSRs, such as higher efficiency of amplification, a gene-associated nature (and hence potential to correlate with functional sequence variation) and enhanced cross-species transferability (Barbara et al. 2007; Kaur et al. 2011, Kaur et al. 2012). For the EST–SNP markers, a lower rate of validation (75 %) was observed in the present study than in a previous report (90.75 %) (Gaur et al. 2012) which could be due to differences in methods used for SNP calling between the two studies.
Prior reports of limited intraspecific genetic diversity are consistent with results from the present study, in which a frequency of 0.775 SNPs per kb was observed. Even lower values were obtained from a comparative study between transcriptomes, of 0.043 SNPs per kb for a Kabuli/Desi cultivated chickpea comparison, and 0.821 SNPs per kb for a Kabuli/non-domesticated chickpea comparison (Agarwal et al. 2012). All of these values are markedly lower than estimates derived from other cultivated species, such as eucalypt (62.5 SNPs per kb, Kulheim et al. 2009), perennial ryegrass (18.5 SNPs per kb, Cogan et al. 2006), rice (6.8 SNPs per kb, Subbaiyan et al. 2012), maize (5 SNPs per kb, Gore et al. 2009) and soybean (2.7 SNPs per kb, Choi et al. 2007). These factors suggest that a large resource of molecular genetic marker assays is required to identify substantial numbers of sequence polymorphisms that will be polymorphic in any given cross. The use of genic sequences, such as ESTs, as templates for resequencing can efficiently address these requirements. It may also be interesting to speculate about the narrow genetic diversity within cultivated chickpea genome being linked to more variation in the promoter regions of the genes. Such an activity would require exploitation of the draft whole genome sequences (Jain et al. 2013, Varshney et al. 2013).
Attributes of genetic linkage maps
Subsets of EST-derived SSRs and SNPs were further used for linkage analysis, to generate chickpea intraspecific genetic maps with, to the best of our knowledge, the highest currently available genic marker density (Flandez-Galvez et al. 2003a, b; Radhika et al. 2007; Gaur et al. 2011). The total lengths of each genetic map are comparable to those from previous studies of chickpea (Gaur et al. 2011, 2012; Thudi et al. 2011). In the current study, a total of 337 publicly available SSRs were included in screening of mapping populations, and were critical for assignment of LGs (Gaur et al. 2012, 2011; Thudi et al. 2011). In those instances in which no such markers were represented on a given LG, attribution could generally be inferred through cross-comparison of the two maps generated in the current study. High-resolution genetic maps based on gene-based markers are useful for many applications apart from basic trait dissection, such as cross-inference of QTL identity between different studies, candidate gene selection, development of diagnostic markers for important agronomic traits, and ordering of genome sequence scaffolds into pseudomolecules. Linkage maps have previously been used for anchoring and orientation of scaffolds in whole genome sequencing projects for many crop species including soybean (Hyten et al. 2010), watermelon (Ren et al. 2012), grape (Jaillon et al. 2007) and cucumber (Huang et al. 2009). The linkage maps described here would be highly useful for future improvements to the chickpea genome assembly.
Conserved macrosynteny between chickpea and M. truncatula
Synteny between genomes can facilitate the transfer of genetic information between closely related crops, the efficacy of which depends on the degree of conservation of gene order and content. Several prior studies have reported syntenic relationships based on genetic map comparisons between different legume species such as M. truncatula (Choi et al. 2004), cowpea (Muchero et al. 2009), peanut (Bertioli et al. 2009) and common bean (Galeano et al. 2011). However, due to the historical underdevelopment of the chickpea genomic resource, comparable information has been limited until recently. Comparison of sequences underpinning SNP markers that were assigned to both linkage maps in the present study revealed substantial macrosynteny with the genome of M. truncatula, consistent with other recent studies (Hiremath et al. 2012; Varshney et al. 2013). In most instances, multiple matches were obtained from each chickpea LG against specific Mt chromosomes, indicative of evolutionary translocations within the respective lineages of the Galegoid (cool-season) legumes. For example, chickpea LG2 was highly colinear with MtChr5, but some marker loci were more obviously related to MtChr2. Similarly, blocks of conserved synteny were observed between LG4 of chickpea and MtChr1, but some of the more distal markers showed matches to MtChr3 and MtChr5. The latter observation suggests that chromosomal rearrangements may have preferentially occurred towards the telomeric ends of chromosomes over evolutionary time, as also observed in other similar studies from various plant species (Jones et al. 2002; McLean et al. 2010). All of the results obtained in this study were found to be highly concordant with the syntenic relationship study performed by Varshney et al. (2013), in which a large number of extended (>10 kb) conserved syntenic blocks were reported between the M. truncatula and chickpea genomes. This information will be highly valuable for a candidate gene selection approach in order to develop gene-based markers for different traits of interest in chickpea, as well as to understand the evolutionary history of this species relative to those of other legumes.
Identification of QTLs controlling ascochyta blight resistance
Molecular analysis revealed a total of three QTLs conferring resistance to ascochyta blight across different parental sources. CIM identified two distinct QTLs to be associated with ascochyta blight resistance in the Lasseter × ICC3996 population, ab_QTL1 on LG4.1 and ab_QTL2 on LG4.2, and one QTL in S95362 × Howzat. Frequency histograms indicated the presence of multiple genes in both mapping populations, but the detection of only one QTL in S95362 × Howzat could be due to the size of the population used for mapping and QTL analysis, which could affect the number of recombination events and hence the detection of genomic regions. Moreover, Howzat is a moderately susceptible cultivar in comparison to Lasseter. Therefore, levels of resistance were higher in the S95362 × Howzat population compared to Lasseter × ICC3996, providing another possible reason for the inability to detect ab_QTL2. Both mapping populations were subjected to phenotypic assessment at the same time within each trial year using the same pathogen inoculum (a field-derived mixture of isolates of A. rabiei). However, differences were observed between V p proportions accounted for by apparently common QTLs between the two trial years, potentially due to differences in the environmental conditions between two time intervals. Although abQTL_1 varies in size and total marker content between the two populations, the presence of common flanking markers indicates the same genomic region.
One of the two QTLs (ab_QTL1) identified in the present study is coincident with previously published QTLs, based on common linked marker loci (Flandez-Galvez et al. 2003a, Udupa and Baum 2003, Lichtenzveig et al. 2006, Anbessa et al. 2009). In order to test the possibility that ab_QTL2 is coincident with the previously described QTL of similar location described by Madrid et al. (2012), an attempt was made to empirically map the CaETR-1a/b and NCPGR91 markers that flank the latter QTL, as well as the adjacent GAA47 marker. However, no polymorphism was detected between the mapping family parents. Due to the probabilistic nature of QTL mapping, it is formally possible that the two QTLs are indeed identical. However, as sequences underpinning the ab_QTL2 linked-SNP loci matched regions of both the M. truncatula (Mtchr 7, c. 22.5 Mbp) and chickpea genomes (Cachr4, c. 12.5–13.5 Mbp) that are distinct from those detected by the CaETR-1a/b and NCPGR91 genes (Mtchr1, c. 19.9 Mbp; Cachr4, 1.6–4.5 Mbp), it is also possible that ab_QTL2 is novel to the present study.
The observation here of two QTLs on LG4 for Lasseter × ICC3996 is hence in accordance with previous studies, which reported 2–3 interacting QTLs on LG4, supporting the idea that a cluster of ascochyta blight resistance genes may exist in this genomic region (Anbessa et al. 2009). Other studies have also identified additional QTLs on other LGs (LG2, 3, 6 and 8) which accounted for a larger proportion of genetic variation compared to the current study. This variation could be due to various factors, such as use of different genetic material, different methodology, glasshouse versus field evaluation and interspecific versus intraspecific crosses. These results can be further validated under field conditions and can be directly applied to chickpea breeding programs.
Three candidate genes associated with plant defence mechanisms were identified in the QTL-containing genomic intervals (ab_QTL1). These genes displayed functional annotations as hevamine-A-like gene and acidic endochitinase-like genes, the former being highly similar to a M. truncatula chitinase. Chitinase-like genes have been shown to be mediate a well-established defence response in chickpea by inhibiting fungal growth of the invading fungus by cell wall digestion (Jayakumar et al. 2005), and are hence plausible candidates for QTL function.
Implications in chickpea breeding programs
The development of genetic linkage maps based on gene-based markers has facilitated the identification of genomic regions underlying ascochyta blight resistance in chickpea. Genetic markers flanking the QTL-containing regions identified in this study are capable of further validation in a diverse set of C. arietinum germplasm, enabling a marker-assisted selection approach for introgression of such regions derived from parental germplasm chickpea breeding programs. Although the current study reports two genomic regions conferring resistance to ascochyta blight, some additional sources of resistance have been reported in the literature and their relevance to Australian chickpea breeding programs still needs to be understood. As the chickpea reference genome sequence has recently become available, sequence analysis in the vicinity of QTLs will permit development of additional SNP loci to allow the selection of linked markers across a broad range of germplasm sources. A gene pyramiding approach is desired to build durable resistance such that the crop will be safeguarded against the ascochyta blight pathogen. Finally, integration of genetic mapping and phenotypic evaluation studies conducted by several research groups will also provide a better understanding of the genetic basis of ascochyta blight resistance.
References
Agarwal G, Jhanwar S, Priya P, Singh VK, Saxena MS et al (2012) Comparative analysis of kabuli chickpea transcriptome with desi and wild chickpea provides a rich resource for development of functional markers. PLoS ONE 7(12):e52443. doi:10.1371/journal.pone.0052443
Anbessa Y, Taran B, Warkentin TD, Tullu A, Vandenberg A (2009) Genetic analyses and conservation of QTL for ascochyta blight resistance in chickpea (Cicer arietinum L.). Theor Appl Genet 119(4):757–765
Barbara T, Palma-Silva C, Paggi GM, Bered F, Fay MF, Lexer C (2007) Cross-species transfer of nuclear microsatellite markers: potential and limitations. Mol Ecol 16(18):3759–3767
Bertioli DJ, Moretzsohn MC, Madsen LH, Sandal N, Leal-Bertioli SC, Guimaraes PM, Hougaard BK, Fredslund J, Schauser L, Nielsen AM et al (2009) An analysis of synteny of Arachis with Lotus and Medicago sheds new light on the structure, stability and evolution of legume genomes. BMC Genomics 5:16
Buhariwalla HK, Jayashree B, Eshwar K, Crouch JH (2005) Development of ESTs from chickpea roots and their use in diversity analysis of the Cicer genus. BMC Plant Biol 5:16
Chabane K, Ablett GA, Cordeiro GM, Valkoun J, Henry RJ (2005) EST versus genomic derived microsatellites for genotyping wild and cultivated barley. Genet Res Crop Evol 52:903–909
Choi HK, Kim DJ, Uhm T, Limpens E, Lim H, Mun JH, Kalo P, Penmesta RV, Seres A, Kulikova O et al (2004) A sequence based genetic map of Medicago truncatula and comparison of marker colinearity with M. sativa. Genetics 166:1463–1502
Choi I-Y, Hyten DL, Matukumalli LK, Song Q, Chaky JM, Quigley CV, Chase K, Lark KG, Reiter RS, Yoon M-S, Hwang E-Y, Yi S-I et al (2007) A soybean transcript map: gene distribution, haplotype and single-nucleotide polymorphism analysis. Genetics 176:685–696
Choudhary S, Sethy NK, Shokeen B, Bhatia S (2009) Development of chickpea EST-SSR markers and analysis of allelic variation across related species. Theor Appl Genet 118(3):591–608
Choudhary S, Gaur R, Gupta S (2012a) EST-derived genic molecular markers: development and utilization for generating an advanced transcript map of chickpea. Theor Appl Genet 124(8):1449–1462
Choudhary P, Khanna SM, Jain PK, Bharadwaj C, Kumar J, Lakhera PC, Srinivasan R (2012b) Genetic structure and diversity analysis of the primary gene pool of chickpea using SSR markers. Genet Mol Res 11(2):891–905
Cogan NOI, Ponting RC, Vecchies A, Drayton MC, George J, Dracatos PM, Dobrowlski M, Sawbridge TI et al (2006) Gene-associated single nucleotide polymorphism discovery in perennial ryegrass (Lolium perenne L.). Mol Gen Genet 276:101–112
Cong B, Liu JP, Tanksley SD (2002) Natural alleles at a tomato fruit size quantitative trait locus differ by heterochronic regulatory mutations. Proc Natl Acad Sci USA 99(21):13606–13611
Flandez-Galvez H, Ford R, Pang EC, Taylor PW (2003a) An intraspecific linkage map of the chickpea (Cicer arietinum L.) genome based on sequence ged microsatellite site and resistance gene analog markers. Theor Appl Genet 106(8):1447–1456. doi:10.1007/s00122-003-1199-y
Flandez-Galvez H, Ades PK, Ford R, Pang ECK, Taylor PWJ (2003b) QTL analysis for ascochyta blight resistance in an intraspecific population of chickpea (Cicer arietinum L.). Theor Appl Genet 107:1257–1265
Galeano CH, Fernandez AC, Franco-Herrera N, Cichy KA, McLean PE, Vanderleyden J, Blair MW (2011) Saturation of an intra-gene pool linkage map: towards a unified consensus linkage map for fine mapping and synteny analysis in common bean. PLoS ONE 6:e28135
Garg R, Patel RK, Tyagi AK, Jain M (2011) De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res 18(1):53–63
Gaur R, Sethy NK, Choudhary S, Shokeen B, Gupta V, Bhatia S (2011) Advancing the STMS genomic resources for defining new locations on the intraspecific genetic linkage map of chickpea (Cicer arietinum L.). BMC Genomics 12:117
Gaur R, Azam S, Jeena G, Khan AW, Choudhary S, Jain M, Yadav G, Tyagi AK, Chattopadhyay D, Bhatia S (2012) High-throughput SNP discovery and genotyping for constructing a saturated linkage map of chickpea (Cicer arietinum L.). DNA Res 19(5):357–373
Gore MA, Wright MH, Ersoz ES, Bouffard P, Szekeres ES, Jarvie TP, Hurwitz BL, Narechania A, Harkins TT, Grills GS et al (2009) Large-scale discovery of gene-enriched SNPs. Plant Genome 2:121–133
Gujaria N, Kumar A, Dauthal P, Dubey A, Hiremath P, Prakash AB, Farmer A, Bhide M, Shah T, Gaur PM, Upadhyaya HD, Bhatia S, Cook DR, May GD, Varshney RK (2011) Development and use of genic molecular markers (GMMs) for construction of a transcript map of chickpea (Cicer arietinum L.). Theor Appl Genet 122(8):1577–1589
Hiremath PJ, Farmer A, Cannon SB, Woodward J, Kudapa H, Tuteja R, Kumar A, Bhanuprakash A, Mulaosmanovic B, Gujaria N, Krishnamurthy L, Gaur PM, Kavikishor PB, Shah T, Srinivasan R, Lohse M, Xiao Y, Town CD, Cook DR, May GD, Varshney RK (2011) Large-scale transcriptome analysis in chickpea (Cicer arietinum L.), an orphan legume crop of the semi-arid tropics of Asia and Africa. Plant Biotechnol J 9(8):922–931
Hiremath PJ, Kumar A, Penmetsa RV, Farmer A, Schlueter JA, Chamarthi SK, Whaley AM, Carrasquilla-Garcia N, Gaur PM, Upadhyaya HD, Kavi Kishor PB, Shah TM, Cook DR, Varshney RK (2012) Large-scale development of cost-effective SNP marker assays for diversity assessment and genetic mapping in chickpea and comparative mapping in legumes. Plant Biotechnol J 10(6):716–732
Huang S, Li R, Zhang Z, Li L, Gu X, Fan W, Lucas WJ, Wang X, Xie B, Ni P, Ren Y, Zhu H, Li J, Lin K, Jin W, Fei Z, Li G, Staub J, Kilian A, van der Vossen EAG, Wu Y, Guo J, He J, Jia Z, Ren Y, Tian G, Lu Y, Ruan J, Qian W, Wang M, Huang Q, Li B, Xuan Z, Cao J, Asan WuZ, Zhang J, Cai Q, Bai Y, Zhao B, Han Y, Li Y, Li X, Wang S, Shi Q, Liu S, Cho WK, Kim J-Y, Xu Y, Heller-Uszynska K, Miao H, Cheng Z, Zhang S, Wu J, Yang Y, Kang H, Li M, Liang H, Ren X, Shi Z, Wen M, Jian M, Yang H, Zhang G, Yang Z, Chen R, Liu S, Li J, Ma L, Liu H, Zhou Y, Zhao J, Fang X, Li G, Fang L, Li Y, Liu D, Zheng H, Zhang Y, Qin N, Li Z, Yang G, Yang S, Bolund L, Kristiansen K, Zheng H, Li S, Zhang X, Yang H, Wang J, Sun R, Zhang B, Jiang S, Wang J, Du Y, Li S (2009) The genome of the cucumber Cucumis sativus L. Nat Genet 41(12):1275–1281
Hyten DL, Cannon SB, Song Q, Weeks N, Fickus EW, Shoemaker RC, Specht JE, Farmer AD, May GD, Cregan PB (2010) High-throughput SNP discovery through deep resequencing of a reduced representation library to anchor and orient scaffolds in the soybean whole genome sequence. BMC Genomics 11:38
Iruela M, Castro P, Rubio J, Cubero JI, Jacinto C, Millan T, Gil J (2007) Validation of a QTL for resistance to ascochyta blight linked to resistance to fusarium wilt race 5 in chickpea (Cicer arietinum L.). Eur J Plant Pathol 119:29–37
Jaillon O, Aury J-M, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C, Vezzi A, Legeai F, Hugueney P, Dasilva C, Horner D, Mica E, Jublot D, Poulain J, Bruyere C, Billault A, Segurens B, Gouyvenoux M, Ugarte E, Cattonaro F, Anthouard V, Vico V, Del Fabbro C, Alaux M, Di Gaspero G, Dumas V, Felice N, Paillard S, Juman I, Moroldo M, Scalabrin S, Canaguier A, Le Clainche I, Malacrida G, Durand E, Pesole G, Laucou V, Chatelet P, Merdinoglu D, Delledonne M, Pezzotti M, Lecharny A, Scarpelli C, Artiguenave F, Pe ME, Valle G, Morgante M, Caboche M, Adam-Blondon A-F, Weissenbach J, Quetier F, Wincker P, French-Italian P (2007) The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449(7161):463–467
Jain M, Misra G, Patel RK, Priya P, Jhanwar S, Khan AW, Shah N, Singh VK, Garg R, Jeena G, Yadav M, Kant C et al (2013) A draft genome sequence of the pulse crop species chickpea (Cicer arietinum L.). Plant J 74(5):715–729. doi:10.1111/tpj.12173
Jayakumar P, Gan PY, Gossen BD, Warkentin TD, Banniza S (2005) Ascochyta blight of chickpea: infection and host resistance mechanisms. Can J Plant Pathol 27:499–509
Jhanwar S, Priya P, Garg R, Parida SK, Tyagi AK, Jain M (2012) Transcriptome sequencing of wild chickpea as a rich resource for marker development. Plant Biotechnol J 10(6):690–702
Jones ES, Mahoney NL, Hayward MD, Armstead IP, Jones JG, Humphreys MO, King IP, Kishida T, Yamada T et al (2002) An enhanced molecular marker based genetic map of perennial ryegrass (Lolium perenne) reveals comparative relationships with other Poaceae genomes. Genome 45:282–295
Kaur S, Cogan NOI, Pembleton LW, Shinozuka M, Savin KW, Materne M, Forster JW (2011) Transcriptome sequencing of lentil based on second-generation technology permits large-scale unigene assembly and SSR marker discovery. BMC Genomics 12:265
Kaur S, Pembleton L, Cogan N, Savin K, Leonforte T, Paull J, Materne M, Forster J (2012) Transcriptome sequencing of field pea and faba bean for discovery and validation of SSR genetic markers. BMC Genomics 13(1):104
Kosambi DD (1944) The estimation of map distances from recombination values. Ann Eugen 12:172–175
Kulheim C, Yeoh SH, Maintz J, Foley WJ, Moran GF (2009) Comparative SNP diversity among four Eucalyptus species for genes from secondary metabolite biosynthetic pathways. BMC Genomics 24:452
Labdi M, Robertson LD, Singh KB, Charrier A (1996) Genetic diversity and phylogenetic relationships among the annual Cicer species as revealed by isozyme polymorphism. Euphytica 88(3):181–188
Lichtenzveig J, BonWl DJ, Zhang HB, Shtienberg D, Abbo S (2006) Mapping quantitative trait loci in chickpea associated with time to Xowering and resistance to Didymella rabiei the causal agent of ascochyta blight. Theor Appl Genet 113:1357–1369
Madrid E, Rajesh PN, Rubio J, Gil J, Millan T, Chen W (2012) Characterization and genetic analysis of an EIN4-like sequence (CaETR-1) located in QTL(AR1) implicated in ascochyta blight resistance in chickpea. Plant Cell Rep 31:1033–1042
Madrid E, Chen W, Rajesh PN, Castro P, Millan T, Gil J (2013) Allele-specific amplification for the detection of ascochyta blight resistance in chickpea. Euphytica 189:183–190
Manly KF, Cudmore RH, Meer JM (2001) Map manager QTX, cross-platform software for genetic mapping. Mamm Genome 12:930–932
Mattioni C, Cherubini M, Taurchini D, Villani F, Martin MA (2010) Evaluation of genomic SSRs and EST-SSRs markers in genetic diversity studies in European chestnut populations. Acta Hort 866:151–156
McLean P, Mamidi S, McConnell M, Chikara S, Lee R (2010) Synteny mapping between common bean and soybean reveals extensive blocks of shared loci. BMC Genomics 11:184
Moreno MT, Cubero JI (1978) Variation in Cicer arietinum L. Euphytica 27:465–485
Muchero W, Diop NN, Bhat PR, Fenton RD, Wanamrker S, Pottorff M, Hearne S, Cisse N, Fatokun C, Ehlers JD, Roberts PA, Close TJ (2009) A consensus genetic map of cowpea [Vigna unguiculata (L) Walp.] and synteny based on EST-derived SNPs. Proc Natl Acad Sci USA 106:18159–18164
Nayak SN, Zhu H, Varghese N, Datta S, Choi HK, Horres R, Jungling R, Singh J, Kishor PB, Sivaramakrishnan S, Hoisington DA, Kahl G, Winter P, Cook DR, Varshney RK (2010) Integration of novel SSR and gene-based SNP marker loci in the chickpea genetic map and establishment of new anchor points with Medicago truncatula genome. Theor Appl Genet 120:1415–1441
Radhika P, Gowda SJ, Kadoo NY, Mhase LB, Jamadagni BM, Sainani MN, Chandra S, Gupta VS (2007) Development of an integrated intraspecific map of chickpea (Cicer arietinum L.) using two recombinant inbred line populations. Theor Appl Genet 115:209–216
Ren Y, Zhao H, Kou Q, Jiang J, Guo S, Zhang H, Hou W, Zou X, Sun H, Gong G, Levi A, Xu Y (2012) A high resolution genetic map anchoring scaffolds of the sequenced watermelon genome. PLoS ONE 7:e29453
Schuelke M (2000) An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol 18:233–234
Sefera T, Abebie B, Gaur PM, Assefa K, Varshney RK (2011) Characterisation and genetic diversity analysis of selected chickpea cultivars of nine countries using simple sequence repeat (SSR) markers. Crop Pasture Sci 62:177–187
Singh KB, Hawtin GC, Nene YL, Reddy MV (1981) Resistance in chickpeas to Ascochyta rabiei. Plant Dis 65:586–587
Subbaiyan GK, Waters DLE, Katiyar SK, Sadananda AR, Vaddadi S, Henry RJ (2012) Genome-wide DNA polymorphisms in elite indica rice inbreds discovered by whole-genome sequencing. Plant Biotechnol J 10:623–634
Thudi M, Bohra A, Nayak SN, Varghese N, Shah TM, Penmetsa RV, Thirunavukkarasu N, Gudipati S, Gaur PM, Kulwal PL, Upadhyaya HD, Kavikishor PB, Winter P, Kahl G, Town CD, Kilian A, Cook DR, Varshney RK (2011) Novel SSR markers from BAC-end sequences, DArT arrays and a comprehensive genetic map with 1,291 marker loci for chickpea (Cicer arietinum L.). PLoS ONE 6:e27275
Udupa SM, Baum M (2003) Genetic dissection of pathotype-specific resistance to ascochyta blight disease in chickpea (Cicer arietinum L.) using microsatellite markers. Theor Appl Genet 106:1196–1202
van der Knaap E, Tanksley SD (2003) The making of a bell pepper-shaped tomato fruit: identification of loci controlling fruit morphology in yellow stuffer tomato. Theor Appl Genet 107:139–147
Varshney RK, Hiremath PJ, Lekha P, Kashiwagi J, Balaji J, Deokar AA, Vadez V, Xiao Y, Srinivasan R, Gaur PM, Siddique KHM, Town CD, Hoisington DA (2009) A comprehensive resource of drought- and salinity-responsive ESTs for gene discovery and marker development in chickpea (Cicer arietinum L.). BMC Genomics 10:523
Varshney RK, Song C, Saxena RK, Azam S, Yu S, Sharpe AG, Cannon S, Baek J, Rosen BD, Taran B et al (2013) Draft genome sequence of chickpea (Cicer arietinum L.) provides a resource for trait improvement. Nat Biotechnol 31:240–248
Voorips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78
Wang S, Basten CJ, Zeng Z-B (2012) Windows QTL Cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh, NC. (http://statgen.ncsu.edu/qtlcart/WQTLCart.htm)
Acknowledgments
This work was supported by funding from the Victorian Department of Environment and Primary Industries and the Grains Research and Development Council, Australia.
Author information
Authors and Affiliations
Corresponding author
Additional information
Amber Stephens and Maria Lombardi have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
11032_2013_9950_MOESM1_ESM.xls
Details of all SSR markers used in linkage mapping analysis: This file contains a list of all SSR marker assays including those that are public and those developed as part of the present study, along with statistics on amplification efficiency and polymorphism rate (XLS 45 kb)
11032_2013_9950_MOESM2_ESM.xls
Sequence information of all of the SSR primer pairs identified and designed using BatchPrimer3: This file contains all of the information (sequence information, orientation, sequence length, expected product length, expected position on Mt genome, Tm, GC content and SSR motif length) on SSR primer pairs designed using BatchPrimer 3 (XLS 2282 kb)
11032_2013_9950_MOESM3_ESM.xls
Details of the 768plex SNP-OPA design: This file contains names and sequence information for all SNP markers used for linkage mapping (XLS 199 kb)
11032_2013_9950_MOESM4_ESM.txt
Consensus sequences of 454 assembled contigs: The data represents the consensus sequences of 20,846 assembled contigs generated as a result of de novo assembly of chickpea ESTs (TXT 17941 kb)
11032_2013_9950_MOESM5_ESM.txt
Sequence information of 454 singletons: The data represents the sequence information on all the singletons generated from de novo assembly of chickpea ESTs. (TXT 21148 kb)
11032_2013_9950_MOESM6_ESM.xls
Characterization of a sub-set of EST-SSRs on wild and cultivated genotypes of chickpea. The table presents data on number and size of alleles amplified from screening of 96 EST-SSR primer pairs on 5 genotypes of chickpea (XLS 22 kb)
11032_2013_9950_MOESM7_ESM.emf
Representative clustering patterns generated by the Illumina GoldenGate® SNP Genotyping assay: The file contains an example of clustering patterns obtained from SNP genotyping assay on two mapping populations. The data point colour codes represent: red, AA (homozygous); blue, BB (homozygous); purple, AB (heterozygous); black, no call (missing data). (EMF 1068 kb)
11032_2013_9950_MOESM8_ESM.xls
Linkage map statistics from Lasseter x ICC3996 and S95362 x Howzat populations: This file contains details of different markers (SSRs and SNPs) and their corresponding positions on different LGs (XLS 80 kb)
11032_2013_9950_MOESM9_ESM.pptx
Comparison between genetic linkage maps of Lasseter x ICC3996 and S95362 x Howzat: This file shows the visual representation of all LGs from both Lasseter x ICC3996 and S95362 x Howzat maps, and the common marker loci between them (PPTX 283 kb)
11032_2013_9950_MOESM10_ESM.pptx
Synteny between genetic linkage maps of Lasseter x ICC3996 and S95362 x Howzat and Medicago truncatula chromosomes: This file shows the visual representation of all LGs from both Lasseter x ICC3996 and S95362 x Howzat maps and their syntenic relationships with Medicago truncatula (PPTX 189 kb)
11032_2013_9950_MOESM11_ESM.jpg
{kind=link}
Frequency distribution of ascochyta blight disease score for two mapping populations: This file contains frequency histograms generated from ascochyta blight disease score from RILs of mapping populations A. S95362 x Howzat in 2005, B. Lasseter x ICC3996 in 2005, C. S95362 x Howzat in 2005, Howzat in 2009 and D. Lasseter x ICC3996 in 2009 (JPEG 150 kb)
11032_2013_9950_MOESM12_ESM.doc
Summary statistics for ascochyta blight resistance QTLs in chickpea: This table represents the data on QTL identification of ascochyta blight resistance in Lasseter x ICC3996 and S95362 X Howzat populations using simple interval mapping (SIM) and composite interval mapping (CIM) (DOC 37 kb)
11032_2013_9950_MOESM13_ESM.xlsx
BLAST analysis of SNP marker sequences flanking QTL regions to chickpea reference genome: This file contains the BLAST analysis data for sequences underpinning SNP loci flanking ab_QTL1 and ab_QTL2 against the chickpea reference genome (XLSX 12 kb)
11032_2013_9950_MOESM14_ESM.docx
Sequences of predicted candidate genes from comparison of flanking markers to reference genome of chickpea: This file contains the sequence information for three candidate genes predicted from the ascochyta blight resistance QTL-containing regions (DOCX 16 kb)
Rights and permissions
About this article
Cite this article
Stephens, A., Lombardi, M., Cogan, N.O.I. et al. Genetic marker discovery, intraspecific linkage map construction and quantitative trait locus analysis of ascochyta blight resistance in chickpea (Cicer arietinum L.). Mol Breeding 33, 297–313 (2014). https://doi.org/10.1007/s11032-013-9950-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11032-013-9950-9