
Abstract
The toxic metabolic product aflatoxin produced by the opportunistic fungus Aspergillus flavus (Link:Fr) in maize (Zea mays L.) can cause disease and economic harm when levels exceed very minute quantities. The selection of resistant germplasm has great potential to reduce the problem, but the highly quantitative nature of the trait makes this a difficult endeavor. The identification of aflatoxin accumulation resistance quantitative trait loci (QTL) from resistant donor lines and the discovery of linked markers could speed this task. To identify marker–trait associations for marker-assisted breeding, a genetic mapping population of F2:3 families was developed from Mp715, a maize inbred line resistant to aflatoxin accumulation, and T173, a susceptible, southern-adapted maize inbred line. QTL, some with large phenotypic effects, were identified in multiple years on chromosomes 1, 3, 5, and 10, and smaller QTL identified in only 1 year were found on chromosomes 4 and 9. The phenotypic effect of each QTL ranged from 2.7 to 18.5%, and models created with multiple QTL could explain up to 45.7% of the phenotypic variation across years, indicating that the variation associated with the trait can be manipulated using molecular markers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Aspergillus flavus (Link:Fr) is a fungus that is endemic in the southern USA and causes ear rot of maize (Zea mays L.) and significant aflatoxin accumulation, particularly in the southern USA (Payne 1992; Widstrom 1996; Windham and Williams 1998). Aflatoxins have carcinogenic, immunosuppressive, and hepatotoxic properties in humans and animals (Castegnaro and McGregor 1998), and the US Food and Drug Administration prohibits interstate commerce of feed grain containing more than 20 ng g−1 aflatoxin (Park and Liang 1993). This leads to severe potential economic losses by farmers with affected grain. Efforts to reduce aflatoxin accumulation in maize have focused on identifying and selecting natural host resistance factors (Busboom and White 2004; Zhang et al. 1999), and several natural sources of resistance have been identified that exhibit significantly reduced aflatoxin accumulation (Robertson-Hoyt et al. 2007; Campbell and White 1995; Scott and Zummo 1988, 1990, 1992; Williams and Windham 2001). Resistance to aflatoxin accumulation can be achieved by reducing the amount of fungus in the grain, reducing the amount of toxin made by the fungus, or both. By measuring the end quantity of toxin in the grain, artificial selection has worked to reduce both simultaneously. However, the quantitative nature of the trait and strong genotype–environment interaction have limited the success of transferring resistance to commercial hybrids (Campbell and White 1995; Hamblin and White 2000). Furthermore, most resistant sources offer poor agronomic performance. Artificial inoculation of maize kernels with the fungus is necessary to produce the toxin for repeatable selection in the field, a slow and expensive process (Zummo and Scott 1989; Windham et al. 2003). Faster and easier methods such as visual screening of grain in the field are not highly correlated with the quantity of toxin in the grain (Windham and Williams 1998).
The use of molecular genetic markers to transfer specific quantitative trait loci (QTL) from resistant breeding lines to current elite (but susceptible) inbreds could greatly speed resistance breeding efforts. For example, markers could be used to introgress identified QTL to elite parents via marker-assisted backcrossing by using markers that can be either cheaply run in a small laboratory, or automated and run very quickly in a larger one; thus all plant breeding programs (large or small) would be able to use the markers to efficiently pyramid and transfer resistant QTL from one or more donor lines into elite maize inbreds. Alternatively, markers could be used to estimate the breeding value of lines based on the magnitude of effects associated with marker alleles for aflatoxin accumulation resistance and other traits of interest (i.e. genomic selection). Challenges include the fact that no QTL have been found to date that explain more than ~20% of the phenotypic variation in any given mapping experiment, and few are consistent across years or locations (Paul et al. 2003; Brooks et al. 2005; Robertson-Hoyt et al. 2007; Warburton et al. 2009).
To aid in the quick and efficient production of new resistant germplasm, information on the trait, markers linked to the trait, and model-based summaries of the joint performance of the QTL (including gene actions and interactions) would be useful. One germplasm line, Mp715, which was developed and released as a source of resistance to aflatoxin accumulation by the USDA-ARS Corn Host Plant Resistance Research Unit (Williams and Windham 2001), possesses a high level of resistance to aflatoxin accumulation, but its late maturity and susceptibility to lodging have limited its use in conventional breeding programs. Molecular marker-assisted selection could facilitate the use of Mp715 in breeding programs.
The goals of this project were: (1) to identify the QTL conferring resistance to aflatoxin accumulation from Mp715; (2) to characterize the magnitude and consistency of resistance in multiple years; and (3) to develop molecular markers to facilitate transfer of identified resistance factors between lines. In addition, by comparing results to previously reported resistance QTL, we hope to improve selection decisions in order to maximize progress in breeding programs for reducing aflatoxin susceptibility in commercial hybrids.
Materials and methods
Population construction and phenotypic evaluation
An F2:3 mapping population was generated from a cross between Mp715 (PI 614819), a breeding line resistant to aflatoxin accumulation (Williams and Windham 2001), and T173 (PI 613179), a susceptible, southern-adapted maize inbred line derived from Mo17 (West et al. 2001). Although Mp715 shows excellent resistance to aflatoxin, it is late maturing and not highly adapted for high yield. Individual F2 plants were selfed to create 250 ear-to-row F2:3 families, each of which was sib-mated to generate sufficient seed for replicated trials. The parents, F1, and 250 F2:3 families were grown in a randomized complete block design with three replications at the R.R. Foil Plant Science Research Center, Mississippi State University, in 2003, 2004, 2005, and 2006 (2003 and 2006 had only two replications each). In 2003, 2004, and 2005, the trial was grown in the same plot, characterized by a Leeper silty clay loam soil, and was planted on April 19, April 22, and April 21, respectively. In 2006, the trial was planted on April 19, in a plot with Marietta fine sandy loam soil. An experimental unit consisted of a 5.1-m single row of each genotype, which was overplanted and then thinned to 20 plants. Management of test plots followed standard agronomic practises adapted to maximize yield.
Aflatoxin accumulation is a commonly used measure of resistance in maize grains (Windham and Williams 1998). Mp715 was a newly identified source of resistance to aflatoxin accumulation at the time this investigation was initiated. Aspergillus flavus strain NRRL 3357 (ATCC #200026) was used in the field evaluations because of its proven ability to generate high levels of aflatoxin in maize grain (Windham and Williams 1998). Fungal inoculum was increased on sterile corn-cob grits (size 2040, Grit-O-Cobs, The Andersons Co., Maumee, OH) in 500-ml flasks, each containing 50 g of grits and 100 ml of sterile, distilled water, and incubated at 28°C for 3 weeks. Conidia in each flask were washed from the grits using 500 ml sterile distilled water containing 20 drops of Tween 20 (Atlas Chemical Industry, Inc., London, UK) per liter and filtered through four layers of sterile cheesecloth. The concentrations of conidia were determined with a hemacytometer and adjusted with sterile distilled water to 9 × 107 conidia per ml. Inoculum not used immediately was refrigerated at 4°C. The top ears on each plant in each plot were inoculated with an A. flavus conidial suspension using the side-needle technique 14 days after mid-silk (50% of the plants in the plot had silks emerged) according to Zummo and Scott (1989). A 3.4-ml suspension containing 3 × 108 conidia ml−1 was injected into each ear through the husk. Ears that were not touching the ground were harvested from plots at maturity, approximately 60 days after mid-silk. Following shelling, samples were ground using a Romer mill (Romer Industries, Inc., Union, MO) according to manufacturer’s instructions. The VICAM AflaTest (VICAM, Watertown, MA) was used to determine aflatoxin concentration in 50-g samples of ground grain from each plot.
Phenotypic data were natural log-transformed to reduce non-normality. Least squares means estimates for missing data were generated using the prediction option of SAS’s Proc GLM (SAS 2003 version, SAS Institute, Cary, NC, USA) fitting an additive model for effects for year, replication, and entry. Broad-sense heritability and the least significant differences in means of parents, F1, and F2 generation aflatoxin accumulation data was also calculated using SAS.
Marker analysis
Leaf tissue samples were collected from each F2 genotype in the population and used to isolate DNA. DNA isolation, genotyping, and linkage analysis were performed using the procedures described by Brooks et al. (2005). Briefly, bulk tissue samples from each F2:3 family were frozen in liquid nitrogen and lyophilized before being ground. DNA extraction was performed by the CTAB method of Saghai-Maroof et al. (1984). More than 400 simple sequence repeat (SSR) markers available from the Maize Genetics and Genomics Database (www.maizegdb.org) and one gene-based marker (peroxidase, mapping to chromosome 7) were screened on the parental lines to identify polymorphisms. Polymorphic markers were genotyped in the entire population, amplified via PCR and visualized with ethidium bromide on 4% (w/v) agarose gels.
Genotypic data were input into the JoinMap mapping software (version 4) (Van Ooijen 2006) to estimate linkage maps for the markers. Linkage groups were constructed using the Maximum Likelihood (ML) mapping. Twenty-five genotypes were excluded from the analysis because of a high level of missing genotypic data, and the remaining 225 genotypes had an overall missing data rate of 2.3%. Composite Interval Mapping (CIM) was performed using QTL Cartographer version 2.5 (Basten et al. 1999) as described by Brooks et al. (2005). To estimate the 0.05 significance threshold for QTL, 1000 permutations were performed with each data set and across all data sets (Doerge and Churchill 1996). Mapping was done separately for each year, and across years. Multiple Interval Mapping (MIM) was also performed to construct a model of the trait variation associated with identified QTL, analyzed jointly, and to test the interactions between pairs of QTL.
Results and discussion
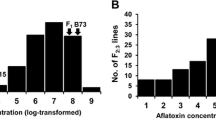
Average aflatoxin scores for the lines in the mapping population at the R.R. Foil Plant Science Research Center were unusually low in 2003, had high similar values in 2004 and 2006, and a slightly higher value in 2005 (Table 1). Standard deviations were high for the untransformed aflatoxin levels (data not shown), which have been reported in past aflatoxin characterization studies as well (Brooks et al. 2005; Campbell and White 1995; Hamblin and White 2000; Paul et al. 2003; Warburton et al. 2009). However, log transformation of the trait considerably reduced the standard deviation, as well as the observed skewness (Table 1). The broad-sense heritability estimate for this trait in one location over 4 years was 0.28, similar to that seen in past experiments (Brooks et al. 2005; Warburton et al. 2009). Average transformed aflatoxin values for both the parents were always significantly different (P < 0.05) from each other and from the values for the F1 and F2 population means except in 2003, where there were no significant differences in any of the values (possibly because all aflatoxin values were reduced). The F1 means were lower than the F2 means, and both were lower than the susceptible parent and higher than the resistant parent in every year except 2003 (Table 2).
Initial screening of SSR markers between the two parents of this linkage population revealed a high (>50%) rate of polymorphic loci (data not shown). A complete linkage map featuring 103 SSR markers distributed across all 10 chromosomes was constructed (Fig. 1). This map correctly reflects known map positions for the markers used (Lawrence et al. 2008), except for one rearrangement on chromosome 2, which may reflect an inversion or incorrect genotyping of the lines. Linkage group 1 could not be resolved as only one group, because short and long arms of this chromosome would not map together in JoinMap unless stringency levels were decreased to the point that other chromosomes began to be grouped together incorrectly.

QTL significantly associated with aflatoxin resistance as measured by the natural log-transformed values of aflatoxin levels in the grain in an F2:3 population derived from Mp715 × T173 cross. Arrows pointing up imply resistance allele contributed by Mp717, and pointing down imply resistance allele contributed by T173. Arrow format refers to the year in which the QTL effect was measured in the MSU field location: arrows with a round dotted line are QTL identified in 2003; a square dotted line are QTL identified in 2004; a dashed line are QTL identified in 2005; a gray solid line are QTL identified in 2005; and a black solid line are QTL identified averaged over all years
QTL were identified by CIM on chromosomes 1, 3 (multiple QTL), 4, 5 (multiple QTL), 9, and 10 (2 QTL) (Fig. 1; Table 3). The QTLs on chromosomes 3 (QTL #1), 5 (QTLs #3 and #8, which are very tightly linked), and 10 (QTL #10) were identified in multiple years. Only 1 minor QTL (on chromosome 10) was not donated by the resistant parent. T173 is a very susceptible line apparently not contributing much to resistance. One QTL (#12) was not significant in any single year, but a detectible peak was present in multiple years; averaged over all years, however, the effect of this QTL became significant (Table 3). The phenotypic variation explained by the QTL ranged from 2.7 to 18.5%, and the models including all the significant QTL explained from 25.4% (in 2005) to 45.7% (over all years) (Table 3).
Based on location on the chromosomes, there were potentially 12 different QTL identified in this study. Only three of these QTL seem to co-locate with QTL reported from other donor lines in the past, including QTL #1 on chromosome 3.06 which may be the same one reported by Paul et al. (2003); QTL #2 on 4.06, which could be the same as the one reported by Brooks et al. (2005); and QTL #3 on 5.05, reported in both of those studies. This suggests that there are at least nine unique QTL for aflatoxin accumulation resistance in the donor line Mp715 that have not been previously reported. This is more resistance QTL for aflatoxin accumulation than has typically been reported in other donor lines in past mapping experiments, which indicates the importance of Mp715 as a donor line.
Past QTL mapping experiments of aflatoxin resistance in maize have typically been done in multiple locations and years (Brooks et al. 2005; Paul et al. 2003; Warburton et al. 2009). These studies typically find QTL identified in only 1 year and location, or identified in common over years but not often across locations. Therefore, the high clustering of QTL seen in the present study (Fig. 1) was not unexpected, since all phenotyping was done at one location over 4 years (and in three of the 4 years the trial was grown in the same plot). In addition, the planting dates only ranged from the 19th to the 22nd of April. However, the rainfall and temperature patterns were very different each year. 2003 experienced cooler than normal temperatures for the deep south and heavy rainfall right after planting that continued throughout the entire growing season, conditions that generally promote optimal maize growth. Since aflatoxin is often produced in stressed plant environments, this may be why very little aflatoxin is seen in this year compared to others. 2004 also saw cooler than normal temperatures and some rainfall following planting and for the first 2 months after, but was very dry during grain filling, which may have stressed the growing grains and triggered aflatoxin production by the fungus. 2005 was cool and very dry during planting, but fairly wet and warmer than normal in the last half of the growing season, possibly leading to a humid mature ear promoting fungal growth. 2006 was very dry the entire summer, with higher than usual temperatures, conditions known to promote high aflatoxin production. In fact, higher aflatoxin levels were expected in 2006 compared to 2004 and 2005 due to lower rainfall and the fact that the 2006 trial was grown in sandier soil that should have caused the plants to dry out sooner; however, this was not the case, highlighting some of the difficulties in breeding for aflatoxin resistance in maize.
The model built by the Multiple Interval Mapping method for Trait 1 (2003 aflatoxin values) using all QTL found at LOD 2.4 or higher included five QTL on chromosomes 3, 4, 5, 9, and 10 (Table 3). The total variation explained by these QTL ranged from 3.1 to 15.0%. Partly due to the fact that all these QTL were unlinked, the correlation between QTL was low, ranging from less than 0.0001 to a high of 0.13 (data not shown). Most of the gene action was additive, although some QTL showed dominant gene action, especially in 2003. Many of the additive QTL did also show some dominance effects, but the dominance effect on the trait was usually much lower than the additive, often by an order of magnitude or more. Furthermore, none of the dominance effects was significant at the LOD score threshold of 2.4, so are not shown in Table 3. Partial dominance may be playing a role in the expression of the trait, however, and may explain why the F1 means are closer to the resistant parent than they are to the susceptible parent (Table 2). Some QTL epistatic effects could be measured in every year of this study and are reported in Table 3; however, most interaction effects were very small and always much lower than the main effects. Additive by additive interactions explained more of the variation, which is to be expected as most significant gene actions were additive, but dominant by dominant were more common, although explaining an order of magnitude less of the phenotypic variation, and two dominant by additive interactions were found as well (explaining even less variation). The 5-QTL model accounted for a total of 32% of the genetic variation (Table 3). The models for years 2004–2006 and for the average effects over all years are also reported in Table 3. Some of the QTL overlap, and some are unique to each year, but the interactions for each year follow similar trends.
This study identified two linked QTL on chromosome 5 (#3 and #8), one or both of which was expressed every year and accounted for 9–18.5% of the phenotypic variation. Because these two QTL are closely linked and come from Mp715, this chromosomal region would be an ideal candidate for marker-assisted selection. It is hoped that this QTL will also be stable across geographic locations since it was stable across the range of environments represented by the various years. However, if it is not, or if it is smaller in other locations, more QTL would be needed to stably improve this trait in order to develop cultivars suitable for many different growing locations. Over all years, 12 QTL were identified in a model that will account for 45.7% of the phenotypic variation, and having so many increases the likelihood that at least some of them will confer resistance in other locations. However, 12 QTL would be difficult to work into a marker-assisted breeding program, as one would not expect to see individual progeny possessing the desired (donor) allele for all 12 QTL in a cross between resistant and susceptible lines unless extremely high numbers of progeny were screened. This would not be practical considering the magnitude of the QTL reported here.
Whole genome selection with many random markers across the genome is becoming popular for the improvement of highly quantitative traits in crops and animals with good marker resources; however, because of the non-elite background of this donor line, this method is not expected to work well for transferring this trait into elite backgrounds. The QTL of the largest phenotypic effects and highest LOD scores will therefore be used to create Near Isogenic Lines (NILs) with highly elite inbred lines to confirm the phenotypic effects of each. The NILs containing confirmed QTL will be excellent breeding lines for quick marker-assisted introgression of the QTL into inbred parents of resistant hybrid varieties. Widely studied quantitative traits in maize, including resistance to aflatoxin accumulation, appear to follow the infinitesimal model of genetic control much more closely than was hoped with the advent of molecular markers. Indeed, for flowering time (a quantitative trait once thought not to be so complicated) a fine-scale mapping study using a joint QTL and association mapping approach and very large population size found that nearly 40 QTL contribute to the trait, and none explain more than 5% of the phenotypic variation (Buckler et al. 2009). Therefore, in a diverse, outbreeding species such as maize, incorporating traits via marker-assisted selection has not proven to be simple, as generally multiple QTL must be used, and QTL tend to be identified in (and specific for) only one environment. Lines containing these QTL grown in a very different environment may not show high expression of the trait. However, marker-assisted selection in combination with phenotypic selection is still effective for quantitative traits, because more gain per year will be made on average over environments than would have been made via phenotypic selection alone; therefore, the most efficient strategy for achieving maximum gain for selection of quantitative traits must be one incorporating marker-assisted selection (based perhaps on an index) in conjunction with phenotypic selection.
References
Basten CJ, Weir BS, Zeng ZB (1999) QTL cartographer. v. 1.13. North Carolina State University, Raleigh
Brooks TD, Williams WP, Windham GL, Willcox MC, Abbas HK (2005) Quantitative trait loci contributing resistance to aflatoxin accumulation in maize inbred Mp313E. Crop Sci 45:171–174
Buckler ES, Holland JB, Bradbury PJ, Acharya CB, Brown PJ, Browne C, Ersoz E, Flint-Garcia S et al (2009) The genetic architecture of maize flowering time. Science 325:714–718
Busboom KN, White DG (2004) Inheritance of resistance to aflatoxin production and Aspergillus ear rot of corn from the cross of inbreds B73 and Oh516. Phytopathology 94:1101–1115
Campbell KW, White DG (1995) Evaluation of corn genotypes for resistance to Aspergillus ear rot, kernel infection and aflatoxin production. Plant Dis 79:1039–1045
Castegnaro M, McGregor D (1998) Carcinogenic risk assessment of mycotoxins. Rev Med Vet 149:671–678
Doerge RW, Churchill GA (1996) Permutation tests for multiple loci affecting a quantitative character. Genetics 142:285–294
Hamblin AM, White DG (2000) Inheritance of resistance to Aspergillus ear rot and aflatoxin production of corn from Tex6. Phytopathology 90:292–296
Lawrence CJ, Harper LC, Schaeffer ML, Sen TZ, Seigfried TE, Campbell DA (2008) MaizeGDB: the maize model organism database for basic, translational, and applied research. Int J Plant Genom 2008:496957
Park DL, Liang B (1993) Perspectives on aflatoxin control for human food and animal feed. Trends Food Sci Technol 4:334–342
Paul C, Naidoo G, Forbes A, Mikkilineni V, White D, Rocheford T (2003) Quantitative trait loci for low aflatoxin production in two related maize populations. Theor Appl Genet 107:263–270
Payne GA (1992) Aflatoxins in maize. Crit Rev Plant Sci 10:423–440
Robertson-Hoyt A, Betrán J, Payne GA, White DG, Isakeit T, Maragos CM, Molnár TL, Holland JB (2007) Relationships among resistances to Fusarium and Aspergillus ear rots and contamination by fumonisin and aflatoxin in maize. Phytopathology 97:311–317
Saghai-Maroof MA, Soliman KM, Jorgensen RA, Allard RW (1984) Ribosomal DNA spacer length polymorphism in barley: Mendelian inheritance, chromosomal location and population dynamics. Proc Natl Acad Sci USA 81:8014–8018
Scott GE, Zummo N (1988) Sources of resistance in maize to kernel infection by Aspergillus flavus in the field. Crop Sci 28:504–507
Scott GE, Zummo N (1990) Registration of Mp313E parental line of maize. Crop Sci 30:1378
Scott GE, Zummo N (1992) Registration of Mp420 germplasm line of maize. Crop Sci 32:1296
VanOoijen JW (2006) JoinMap® 4, Software for the calculation of genetic linkage maps in experimental populations. Kyazma BV, Wageningen, Netherlands
Warburton ML, Brooks TD, Krakowsky MD, Shan X, Windham GL, Williams WP (2009) Identification and mapping of new sources of resistance to aflatoxin accumulation in maize. Crop Sci 49:1403–1408
West DR, Kincer DR, Graves CR (2001) Registration of T173 parental line of maize. Crop Sci 41:1375
Widstrom NW (1996) The aflatoxin problem with corn grain. Adv Agron 56:219–280
Williams WP, Windham GL (2001) Registration of maize germplasm line Mp715. Crop Sci 41:1374–1375
Windham GL, Williams WP (1998) Aspergillus flavus infection and aflatoxin accumulation in resistant and susceptible maize hybrids. Plant Dis 82:281–284
Windham GL, Williams WP, Buckley PM, Abbas HK (2003) Inoculation techniques used to quantify aflatoxin resistance in corn. J Toxicol Toxin Rev 22:313–325
Zhang Y, Kang MS, Magari R (1999) Genetics of resistance to kernel infection by Aspergillus flavus in maize. Plant Breed 116:146–152
Zummo N, Scott GE (1989) Evaluation of field inoculation techniques for screening maize genotypes against kernel infection by Aspergillus flavus in Mississippi. Plant Dis 73:313–316
Acknowledgments
The authors express their appreciation for the technical assistance of J. A. Haynes and L. T. Owens, critical reviews and suggestions by Martha Wilcox and Rita Mumm, and statistical advice from D. Rowe. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the US Department of Agriculture.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Warburton, M.L., Brooks, T.D., Windham, G.L. et al. Identification of novel QTL contributing resistance to aflatoxin accumulation in maize. Mol Breeding 27, 491–499 (2011). https://doi.org/10.1007/s11032-010-9446-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11032-010-9446-9