
Abstract
Plasmopara viticola causes downy mildew of grapevine, one of the most important diseases in viticulture. Resistance to this oomycete is present in American and Asian Vitis species, while traditional European Vitis vinifera cvs. for wine and table grape production are susceptible. Breeding aims to achieve resistance through introgression, but the molecular mechanisms are still unknown. Therefore, the differential display approach was used to detect grapevine genes involved in defense. P. viticola sporangia were applied to the lower leaf surface of in vitro plants of the resistant Vitis riparia selection ‘Gloire de Montpellier’ and susceptible cv. ‘Riesling’. Controls were treated with sterile water. Messenger RNAs extracted 12 h post infection were subjected to differential display. Seven transcripts appeared specifically induced during the incompatible interaction. Sequencing showed that they build three classes. One of them, named VRP1, represented by three transcripts of almost identical sequence but differing lengths, showed clear homology to resistance genes of the NBS-LRR type from other plants. Northern hybridizations confirmed its elevated expression in the resistant ‘Gloire de Montpellier’. Redundancy of VRP1 PCR products from V. riparia prevented PCR-walking, so the VRP1 genes were isolated from a BAC-library of the resistant cv. ‘Regent’. Three genes matching the original VRP1 sequences were found within a BAC clone carrying a 134,392 bp insertion. These were referred to as VRP1-1, 1-2 and 1-3. They encode proteins of 798, 811 resp. 813 amino acids and exhibit the structure of CC-NBS-LRR resistance genes. They were genetically mapped to linkage group 10.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Plasmopara viticola [(Berk. and Curt) Berl. et De Toni], the causal agent of grapevine downy mildew, is one of the most devastating pathogens in viticulture. This biotrophic oomycete was accidentally introduced from America and first observed in Europe during the nineteenth century. In consequence, repeated use of fungicides during the vegetation period has become inevitable to protect the grower from the risk of severe losses, as the common European scion cultivars (cvs) of grapevine (Vitis vinifera L. ssp. vinifera) are generally susceptible to the disease. The necessity of recurrent fungicide applications raises concerns about environmental safety, either for modern synthetic or older copper-based protectants and promotes resistance breeding.
Sources of resistance to downy mildew can be found in American and Asian wild species of grapevine. Such genetic resources have been used during decades as donors for the traditional cross-breeding of resistant cultivars. A major problem however is the long time required to breed a new cultivar that joins resistance and high wine quality. Several generations of backcrossing to traditional grapevine cultivars are required to re-establish wine quality. Therefore, efforts are undertaken to use molecular analytical approaches to improve the knowledge about the interaction between the grapevine host and the P. viticola pathogen. Although some cytological and molecular aspects of the host/pathogen interaction have already been studied (e.g. Kortekamp and Zyprian 2003; Hamiduzzaman et al. 2005; Kortekamp 2006), the resistance mechanism is not yet understood at molecular level. Quantitative trait analysis identified QTLs with major and minor effects on the resistance to the pathogen (Fischer et al. 2004; Welter et al. 2007). Resistance gene analog (RGA)-markers co-located with the major QTL for resistance to the pathogen were detected (Di Gaspero et al. 2007; Welter et al. 2007). QTL analysis is a widespread technique to detect resistance factors, but has limitations regarding the identification of genomic regions with minor effects on the phenotype. The investigation of gene expression after challenging the host plant with pathogen thus is an additional and complementary way to elucidate genes involved in the resistance response.
In the present investigation a differential display approach was applied to identify differentially expressed mRNAs after challenging the plants with the pathogen. The resistant Vitis riparia selection ‘Gloire de Montpellier’ was used in comparison to the susceptible V. vinifera ssp. vinifera cv. ‘Riesling’. ‘Gloire de Montpellier’ exhibits a typical hypersensitivity resistance response upon infection with P. viticola (Kortekamp and Zyprian 2003). This differential display analysis initiated the isolation, characterization and genetic mapping of a CC-NBS-LRR resistance candidate gene family in grapevine by full sequence analysis of a corresponding 134 kb BAC clone available in a BAC-library from the resistant grapevine cv. ‘Regent’.
Materials and methods
Plant/pathogen material and experimental infection
Axenic cultures of grapevine cvs. ‘Riesling’ (V. vinifera ssp. vinifera) and ‘Gloire de Montpellier’ (V. riparia) were maintained on modified LS medium (Linsmaier and Skoog 1965; Blaich 1977) at 26–28°C and 16 h light (40–50 μE/m2)/day in glass containers (Weck® glasses). The plants were propagated by cuttings and re-transferred onto fresh medium every 8–12 weeks.
P. viticola sporangia were collected by incubating symptomatic leaves from the field over night in a moist chamber at room temperature. Sporangia were carefully recovered, stored at −25°C and checked for their vitality by microscopy. A suspension of 40,000 vital sporangia/ml was deposited in small droplets (35–40 μl) on the lower leaf surface to challenge the plants. For control experiments the same amount of sterile water was applied. Infected or control leaf tissue was cut out at different time intervals after the experimental application by using a sterile 5 mm diameter cork borer. This material was shock-frozen in liquid nitrogen and stored at −70°C until processing.
RNA and DNA preparation
Approximately 100 mg of tissue was used for total RNA extraction according to Chang et al. (1993), with minor modifications as described (Wielgoss and Kortekamp 2006). PolyA+-RNA was prepared using the Oligotex™ system (Qiagen, Hilden, Germany). Genomic DNA from grapevine was extracted from 2 g fresh material (leaves, shoot axes or roots from in vitro-grown plants) as described by Thomas and Scott (1993). Genomic DNA of the fungus was obtained from 30 to 50 mg of sporangia following the procedure of Lee and Taylor (1990) with minor modifications.
Differential display reverse transcription PCR (DDRT-PCR)
Anchor primers complementary to the 3′-poly A end of mRNA consisted of d(T)11 and two selective nucleotides at their 3′-extension (“Anchor primer kit”, Roth, Karlsruhe, Germany). They were combined with random dekamer primers (“DD primer kit 150”, Roth) in trial amplifications after reverse transcription using the “First strand cDNA synthesis kit” (Fermentas, St.Leon-Rot, Germany). PCR assays were run under standard conditions. DDRT-PCR products were loaded onto denaturing 6% polyacrylamide gels and detected by silver staining.
Cloning and sequencing of DDRT-PCR products
Differentially observed DDRT-PCR products were excised from the gel and eluted according to Tseng et al. (1995). The isolated DNA fragments were precipitated with ethanol and recovered in 10 μl of TE buffer. Five microliter of the eluate was re-amplified using the original primers and PCR conditions. The PCR products were cloned using the “Sure Clone® Ligation Kit” (Pharmacia, Freiburg, Germany) and transformed into competent cells (Hanahan 1983) of Escherichia coli DH5α. Plasmid DNA was isolated from recombinant clones using the “High pure Plasmid Isolation Kit” (Roche Diagnostics, Germany), characterized by restriction analysis and sequenced with the “ABI PRISM BigDye™ Terminator Cycle Sequencing Ready Reaction Kit” (PE Applied Biosystems, Weiterstadt, Germany). Data processing employed DNASIS™ for Windows (Hitachi, San Bruno, CA, USA).
Southern and Northern hybridizations
Southern and Northern hybridizations were performed for the three classes of differentially displayed transcripts detected (VRP1, 2 and 3). Specific primer pairs for each group were designed: VRP1f (forward): GGAAGACAACTACACAC, VRP1r (reverse): GTGCAAGCATGAAGCCTC; VRP2f (forward): GTGTATCCCAATGCCACTGCA, VRP2r (reverse): GCCAGCGACTTCGTGCTCAG; VRP3f (forward): CAAGATGCGATGATGATCAC, VRP3r (reverse): GCCCAGAGATATGTGG. These primers were used for PCR-based synthesis of Digoxigenin-dUTP labeled probes for Southern hybridizations. Genomic DNA was cut with restriction endonucleases BamHI, EcoRI, or HindIII (Fermentas, St.Leon-Rot, Germany) and the fragments were resolved on 1.2% agarose (FMC Bioproducts Biozym, Oldendorf, Germany) in TAE buffer (40 mM Tris–Acetate, 2 mM EDTA, pH 7.9). Transfer of the fragments to positively charged nylon membranes (Roche Diagnostics, Germany) was done according to Southern (1975). The DNA was fixed to the membrane by baking for 2 h at 80°C. Prehybridization, hybridization and detection were performed using the “DIG Easy Hyb” system and CDP-Star (Roche Diagnostics, Germany).
Poly A+ mRNA for Northern hybridizations was prepared by binding the molecules to biotin-labeled d(T)20 oligonucleotides (ARK Scientific, Darmstadt, Germany) and purified with Streptavidin-covered metallic beads (Miltenyi Biotech, Bergisch-Gladbach, Germany) using magnetic recovery. The mRNA was resolved on denaturing agarose gels prepared in MOPS buffer (20 mM MOPS, 5 mM sodium acetate, 1 mM EDTA, pH 7.0) containing 2.47% formaldehyde. The RNA was transferred onto Nytran Plus membranes (Schleicher and Schüll, Kassel, Germany) as described above and cross-linked to the membrane with UV light. Probes were labeled with alpha 32P-dCTP (10 mCi/ml resp. 3.7 × 108 Bq/ml) using the “Random primed labeling kit” (Roche Diagnostics, Germany). The filters were prehybridized in Church-buffer (Church and Gilbert 1984) for 30 min and hybridized in the same buffer containing the probe over night at 65°C. The membrane was washed twice for 30 min at 65°C in wash buffer 1 (1 mM EDTA, 5% SDS, 40 mM NaHPO4, pH 7.2), for 10 min at room temperature in wash buffer 2 (1 mM EDTA, 1% SDS, 40 mM NaHPO4, pH 7.2), wrapped in foil and exposed to Kodak MS X-ray film at −70°C. The membranes were re-used for hybridizations after stripping by boiling in 0.5% SDS, cooling down and drying at room temperature. A β-tubulin probe from Arabidopsis thaliana (U. Conrath, University of Kailserslautern) served as a control for even RNA loading.
BAC-library, screening and sequencing of clone B18G20
The VRP1 primer pair was used to screen a BAC (bacterial artificial chromosome)-library constructed from grapevine cv. ‘Regent’ by PCR. This BAC-library has an average insert size of 102.4 kb and a statistical 10.2-fold coverage of the ‘Regent’ genome with 47,616 clones. The library contains a contamination of cpDNA of 3.36% as revealed by hybridizing high density nylon filters carrying 9,216 clones with a Digoxigenin-labeled PCR probe produced with primer pair trnM and rbcL from cpDNA (Demesure et al. 1995). Half of the library (50% HindIII and 50% MboI clones) was organized into three dimensional pools of 6 × 384 clones in microtiter plates, rows and colums through stacks of six 384 microtiter plates each and individual plate pools of 384 recombinant strains. The positive clone carrying the largest ‘Regent’ DNA fragment (app. 134 kb), was submitted to sequencing (MWG Biotech, Ebersberg, Germany). The assembly of the shotgun sequences resulted in six large contigs, which were linked to each other by direct sequencing of PCR products obtained from contig-specific gap-outreaching primers, resulting in a total length of 134,392 bp.
Sequence analysis of clone B18G20
The DNA sequence of clone B18G20 was scanned for putative protein coding regions and splicing sites using “GeneScan” software (HUSAR, Heidelberg Unix Sequence Analysis Resources, http://genome.dkfz-heidelberg.de). ORFs (open reading frames) larger than 100 amino acids were further considered. The predicted protein sequences were analysed by protein–protein blastx comparison performed in the NCBI database (http://www.ncbi.nlm.nih.gov; Altschul et al. 1997). Blastx alignments were used to identify possible genes not detected by “GeneScan”. The “PileUp” program (HUSAR) was employed for multiple sequence alignments. Motif searches were done with the “MotifScan” software available at the Swiss Protein Database (http://www.isb-sib.ch), “ProSite” (http://www.expasy.org) or the algorithms to search conserved domains at NCBI (http://www.ncbi.nlm.nih.gov/Structure). The “Coil” program (http://www.ch.embnet.org/software/COILS_form.html) was used to scan for coiled-coil structures.
Genetic mapping of the resistance candidate genes
Primer pairs were designed for each of the VRP1 resistance candidate genes to map them independently using the cross population of ‘Regent’ × ‘Lemberger’ (Welter et al. 2007). Due to the high similarity of the three coding regions, the primers were designed within an intron. The primer sequences are: VRP1-1F: GGTAAAATATCGTCTTTTCCAT, VRP1-1R: AGCCAATCGAAGATTATTTG, VRP1-2F: TGCATCGAATTAAGTATTGC; VRP1-2R: CTTACATTCGTGTCGTCAAA; VRP1-3F: CCATCGTAGCTTGCTCTTAT, VRP1-3F: GGAATGAAAATGGAGAGACA. The PCR products were separated on SSCP gels (Serdogel, Serva, Heidelberg, Germany) and visualised by silver staining. JoinMap 3.0 software (Van Ooijen and Voorrips 2001) was employed to genetically map these markers.
Results
Differential display of P. viticola challenged grapevines
Enzyme activity assays and cytological investigations had shown that the defense reaction of ‘Gloire de Montpellier’ results in induction of cytoplasmic peroxidase activity with the highest level reached at 10–15 hpi with P. viticola sporangia (Kortekamp 2001). Hence this time point was chosen for RNA extraction from infected and non-infected leaf tissue of the resistant variety ‘Gloire de Montpellier’ and the susceptible cv. ‘Riesling’. The mRNA obtained was reverse transcribed and used in differential display analysis (Liang and Pardee 1992). PCR reactions combining anchor primers with random decamer primers (DDRT-PCR) displayed several differentially expressed transcripts. Those detectable exclusively in infected ‘Gloire de Montpellier’ leaves (and absent in infected or non-infected ‘Riesling’ resp. non-infected ‘Gloire de Montpellier’) were cloned and sequenced. Seven transcripts of 340–850 b lengths met these criteria (Table 1). Some of them showed strong homology with each other (more than 90% sequence identity) and were grouped into classes. The three transcripts composing class 1 (VRP1—V itis resistance to P lasmopara, sizes 660, 570 resp. 850 b) exhibit pronounced similarities to NBS-LRR type resistance genes known from other plant species (Dangl and Jones 2001). The putatively encoded proteins of all three variants possess LRR (leucine rich repeat) domains.
The second class (VRP2), also containing three transcripts (670, 720 resp. 570 b), showed similarity to a gene of unknown function from Arabidopsis thaliana and rice (Oryza sativa). The seventh transcript (VRP3, 425 b) has rather weak similarity to a MADS-box binding protein gene from Silene latifolia of uncertain significance.
Confirmation of the grapevine origin of the differential transcripts
To verify the plant origin of the differential transcripts prepared from RNA present during the interaction of host and pathogen, specific primers were designed for each of the three VRP classes and tested in PCR reactions with genomic DNA from grapevine (cvs. ‘Gloire de Montpellier’, ‘Riesling’, ‘Kerner’, ‘Regent’) and P. viticola. Amplificates of the expected sizes (452 bp for VRP1, 360 bp for VRP2 and 395 bp for VRP3) were obtained from all the Vitis cvs. tested, but never from pathogen DNA (data not shown). Southern hybridization of probes of VRP1, -2 and -3 with genomic DNA of the four grapevine cvs. and P. viticola DNA confirmed the host origin of the cDNAs. Several polymorphic hybridizing bands were observed for VRP1 (up to five HindIII fragments) and VRP2 (up to four HindIII fragments), providing evidence for the presence of small gene families.
Verification of basal and pathogen-induced transcription of VRP1
Northern hybridizations using the identical conditions as in the differential display analysis revealed different expression patterns for VRP1, VRP2 and VRP3. VRP1 showed a higher signal of basal expression under non-infected conditions in the resistant cv. ‘Gloire de Montpellier’ as compared to susceptible ‘Riesling’. In addition, the hybridization signal appeared induced under infection with P. viticola to a significantly higher level as in infected ‘Riesling’ (Fig. 1). In contrast to VRP1, pathogen-induced expression of the other classes of transcripts could not be verified. VRP2 was expressed at very low level and its signal even appeared reduced during infection with the pathogen. A signal hybridizing to the VRP3-specific probe was barely detectable, indicating a very low level of expression (data not shown). Further analysis thus was focussed on VRP1.

Northern-Blots showing the expression of the reference gene ß-Tubulin and the target gene VRP1. The expression was evaluated with mRNA extracted from leaves of ‘Gloire de Montpellier’ (resistant) and ‘Riesling’ (susceptible) 12 h after infection with Plasmopara viticola (Inf.) in comparison to non-infected plants (C = control)
Identification of the full length VRP1 genes in a ‘Regent’ BAC clone
The high sequence similarity to NBS-LRR type resistance genes and the inducibility during P. viticola challenge made the VRP1 transcripts interesting candidates for further studies. Attempts to isolate the full length genes by PCR-walking from ‘Gloire de Montpellier’ genomic DNA remained unsuccessful due to the generation of redundant products. The orthologous VRP1 genes were hence isolated from the P. viticola resistant cv. ‘Regent’ using a comprehensive BAC (bacterial artificial chromosome) library available.
Screening with the original VRP1 primer pair identified six positive clones. The largest one, clone B18G20 carrying 134 kb of ‘Regent’ genomic DNA, was submitted to sequencing. After gap closure of six large contigs assembled from shotgun sequencing a total length of 134,392 bp was obtained (GenBank accession EU 669439).
Genomic organization of BAC clone B18G20
The sequence of BAC clone B18G20 contains ten open reading frames (ORFs) homologous to known proteins. Their position and orientation is shown in Fig. 2. The putative functions of the predicted proteins are listed in Table 2. Half of the ORFs seem to encode proteins of transposable elements (ORFs 1, 3, 4, 8 and 9).

(a) Genetic organisation of the 134,392 bp grapevine genomic segment of BAC B18G20. The positions and orientations of ten ORFs identified with GeneScan software are indicated. The three CC-NBS-LRR resistance candidate genes VRP1 are represented by black arrows. Lined arrows represent ORFs that code for polypeptides showing homology to transposable element proteins. (b) Structure of the predicted VRP1 genes. Exons are represented by black boxes and introns by lines. Intron sizes are indicated above their location. The numbers left and right represent the position of the genes in the BAC B18G20 sequence. The number at the left side represents the first base of the start codon (ATG) and the number at the right side the last base of the stop codon (TGA)
The sequences of ORFs 2, 5 and 10 contain the original VRP1 transcript sequence from the differential display. ORF10 includes the closest matching copy at nucleotides 2,682–3,170 counting from its start codon, the alignment exhibiting five nucleotide exchanges (positions 3045, 3131, 3140, 3161, 3166) not altering the encoded amino acid sequence (in the range of amino acids 584–747 of ORF10). These genes were thus designated VRP1-1 (ORF2), VRP1-2 (ORF5) and VRP1-3 (ORF10).
A polypeptide sharing high homology to two ubiquitin domains arranged in tandem as found in the phosphatidylinositol 4-kinase type II protein from Arabidopsis thaliana (Müller-Roeber and Pical 2002) could be encoded by ORF 6. However this putative gene seems to have undergone major rearrangements through retrotranspositional insertion. The two ubiquitin-like domains appear separated by retrotransposonal sequences (positions 84807–84634, Table 2) and a putative 4-kinase protein domain is missing. ORF 7 encodes a heat shock protein and the polypeptide encoded by ORF 4 has homology to a zinc finger protein. Additional small regions showing similarity to retroelements on DNA sequence level not represented in Fig. 2 were identified by blastx analysis (Table 2, bottom).
Structure of the VRP1 resistance candidate genes
The three VRP1 resistance candidate genes on BAC clone B18G20 share the same orientation (Fig. 2). The lengths of VRP1-1, VRP1-2 and VRP1-3 genes from the start to the stop codons are 4,464, 3,037 and 3,418 bp, respectively. This variation is mainly due to different sizes of intron 1. According to splicing predictions VRP1-1 possesses the longest intron 1 (1,551 nt), followed by VRP1-3 (637 nt) and VRP1-2 (195 nt) (Fig. 2). VRP1-2 and VRP1-3 are spliced into five exons, resulting in predicted polypeptides of 811 and 813 amino acids, respectively. In VRP1-1 the third exon seems to have additional splice sites, leading to a smaller protein of 798 amino acids. The different splicing apparently results from point mutations.
The deduced proteins closely resemble each other. Pairwise comparison showed that VRP1-2 and VRP1-3 are most similar, with 87% amino acid identity, whereas VRP1-1 shares 70% resp. 72% of its amino acids with VRP1-2 and VRP1-3. A significant divergence of VRP1-1 from the other two proteins results from the loss of 24 amino acids, caused by the altered splicing.
All three proteins resemble a long list of NBS-LRR plant resistance genes, of which the tobacco mosaic virus resistance gene NRG1 (N requirement gene 1, Peart et al. 2005) is the closest characterized homolog. VRP1-3 shows 39% amino acid identities with NRG1 at an E-value of 2e−157. Furthermore, the splicing pattern of VRP1-2 and VRP1-3 exactly matches the five exons of NRG1. The three VRP proteins also resemble the Arabidopsis gene product of ADR1 (activated disease resistance 1) mediating resistance to Peronospora parasitica and Erysiphe cichoracearum (Grant et al. 2003) with the best E-value of 3−99 in the case of VRP1-3.
Domains of the VRP1 proteins
The NRG1 gene and other plant resistance genes homologous to the VRP1 variants encode the subclass of CC-NBS-LRR type resistance proteins (Dangl and Jones 2001; Meyers et al. 2003). The NBS-LRR proteins in general are thought to be involved in the recognition of specific pathogen effectors and activation of defense reponses. The subclass of CC-NBS-LRRs has a coiled-coil (CC) domain at the N-terminus followed by the NBS (nucleotide binding site) and an LRR (leucine rich repeat, COG 4886) in the C-terminal region, believed to mediate recognition specificity. Specifically ADR1 contains two subdomains identical to those of protein kinases near the N-terminus, followed by the CC, the NBS and nine LRRs (Grant et al. 2003).
Scans for conserved protein motifs and domains identified the VRP1 encoded proteins clearly as members of the CC-NBS-LRR subclass. The formation of coiled-coil structures at the N-termini of the VRP1 peptides was also indicated by “Coils” software. Amino acid residues unique for the CC-NBS-LRR class of resistance genes (Pan et al. 2000) were detected in the NBS region of the VRP1 peptides. A comparison of the NBS domains of the predicted VRP1 proteins and the characterized resistance proteins NRG1, ADR1 and the Arabidopsis downy mildew resistance gene product RPP5 (Parker et al. 1997) with the typical elements is illustrated in Fig. 3. The last tryptophan of the kinase 2 motif (labeled with an arrow) is found characteristically in the CC-NBS-LRR genes and is absent in RPP5 (Position 114, Fig. 3), a member of the different subclass of TIR-NBS-LRR resistance proteins.

Multialignment showing the homology of predicted VRP1 proteins with the product of previously characterized CC-NBS-LRR resistance genes NRG1, ADR1 and the TIR-NBS-LRR resistance gene RPP5. The region compared corresponds to 300 amino acids of the NBS domain. Amino acids were aligned using the “PileUp” program. Consensus residues above 50% are indicated by shading. The conserved domains kinase 1a, kinase 2, RNBS-B, RNBS-C and GLPL and RNBS-D are labeled. The last tryptophane of the kinase 2 motif is specific for CC-NBS-LRR class of resistance genes and highlighted by an arrow (position 114)
The LRR domains of the VRP1 proteins contain seven repeats. Each unit has 24 amino acids. The individual repeats diverge at variable amino acid positions. In Fig. 4 the five most conserved LRR repeats are compared to each other. Similar to the NBS domain (Fig. 3), the LRR repeats of VRP1-2 and VRP1-3 are pratically identical. Only four amino acid residues differ. The most significant change is detected in the fifth LRR repeat (“e”), wich is located within a β-strand/β-turn domain. In comparison of VRP1-1 to VRP1-2 and VRP1-3, a substitution of 24 amino acids is encountered in VRP1-1.

Amino acid comparison of five strongly conserved LRRs of the VRP1 resistance candidate gene ORFs identified on BAC clone B18G20. The letter in annex to the gene indicates the individual LRR. Conserved residues have a black background. The majority of the LRRs show the conserved core sequence LxxLxxLxxxxCxxLxxLxxxLxx. Alignment of the LRRs was done using the “PileUp” software available in the Husar bioinformatic facilities
Besides these protein domains described for the CC-NBS-LRR class of resistance proteins, searches for conserved domains at NCBI also indicated the presence of an RPW8 domain (pfam 05659) at the N-terminus of VRP1-2, although with lower significance (E-value 0.008) as compared to the other elements described above. The genes RPW8.1 and RPW8.2 from Arabidopsis thaliana Ms-0 mediate broad level resistance to powdery mildew (Xiao et al. 2001) and other biotrophic pathogens. These small proteins are made up of a transmembrane domain followed by a short CC domain and likely associated with the cellular endomembrane system. They are supposed to function rather in general defense than specifically in powdery mildew resistance (Wang et al. 2007). Additional “ProSite” motif searches indicated the presence of the RPW8 domain also in VRP1-1 (amino acids 1-150, score 11.3) as well as in VRP1-2 (amino acid 1–148, score 9.95) but not in VRP1-3. Interestingly, NRG1 and ADR1 were both indicated to possess the RPW8 domain with scores of 14.58 for NRG1 (amino acids 1–151) and 13.21 for the first 149 amino acids of ADR1. (The original A. thaliana proteins RPW8.1 and RPW8.2 scored with 29.24 resp. 23.972 over 148 resp. 153 amino acid residues in these “ProSite” comparisons).
VRP1 orthologs in susceptible V. vinifera
To investigate the evolution and function of the VRP1 genes, their sequences from cv. ‘Regent’ were compared to preliminary contigs from the whole grape genome sequencing project of the P. viticola susceptible inbreeding line of V. vinifera cv. ‘Pinot noir’ (PN40024) (http://www.genoscope.cns.fr/externe/English/Projets/Projet_ML/projet.html). Alignment of the ORFs (including their introns) from ‘Regent’ VRP1 genes with the PN40024 contigs clearly identified orthologous genes in PN40024. These were identified on contigs 18783 (VRP1-1), 18782 (VRP1-2) and 18773 (VRP1-3). Both the organization of the ORFs and their predicted polypeptide sequences are highly similar. Only two amino acid changes at positions 147 (Ser to Tyr) in the CC domain and at 644 (Met to Leu) in the LRR region were detected comparing VRP1-1 and its orthologous gene. Nine amino acid substitutions differed between VRP1-2 and the respective orthologue (two in the CC domain at positions 25 and 127, five scattered allover the NBS domain, one in the LRR region (position 733) and one at position 497 in between the NBS and LRR parts of the presumptive protein). VRP1-3 and the corresponding ORF of contig 18773 differ in 34 amino acids and show a deletion of four amino acids in PN40024. The majority of these changes ocurred at the N-termini within the CC domain. Five, resp. four amino acid replacements ocurred in the NBS and LRR regions.
All VRP1 orthologs seem to be transcribed in V. vinifera, as several significantly matching EST sequences were found in database searches.
The sequence context of VRP1 genes
To further understand the relationships between these genes, the sequence ranges 2,000 bp upstream and 2,000 bp downstream of the ORFs were analysed. The three VRP1 genes from ‘Regent’ were compared to each other. VRP1-2 and -3 share a segment of 135 bp of high similarity (only 7 nt changes) directly preceding the start codon. The same sequence is found shifted 68 bp upstream of the start codon of VRP1-1 due to a small insertion. Further upstream of this region, VRP1-1 differs considerably from the other two genes. VRP1-2 and -3 resemble each other closely over a stretch of 1,260 bp in the upstream sequence. Downstream of the stop codon, the three VRP1 genes share 71 bp of similar nucleotides. VRP1-2 and -3 are very similar overall, although some smaller insertions and deletions are evident.
The sequence context of the VRP1 orthologs in the corresponding PN40024 contigs was found to be quite similar. In the case of contig 18782 only 80 bp could be compared, as the corresponding gene lies at the end of contig.
Genetic mapping of VRP1 genes
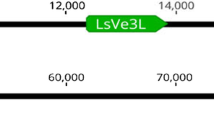
Specific primer pairs were designed to map the three VRP1 genes individually. They were located in the genetic map previously constructed (Welter et al. 2007) employing a segregating population derived from the cross of ‘Regent’ × ‘Lemberger’. The genes were positioned at one extremity of LG 10, 24 cM distant from the microsatellite marker UDV59. No recombination events were found between the three copies using a mapping progeny of 144 individuals.
Discussion
This investigation of the molecular response of the resistant V. riparia selection ‘Gloire de Montpellier’ to P. viticola attack was initiated to increase the knowledge about the genes involved in the incompatible host/pathogen interaction. The differential display approach revealed three different cDNA classes. Two sets of three cDNAs each, classes VRP1 and VRP2, had amplification products of different lengths with very strong sequence similarities (>90% identity) to each other. Southern analysis with VRP1- and VRP2-specific probes revealed small numbers of hybridizing fragments with polymorphic patterns in resistant and susceptible grapevine cvs. This provided evidence that the multiple cDNA bands obtained in DDRT-PCR originated from small gene families and resulted from the transcription of several closely related genes. Partial mispriming (Zhao et al. 1995) or incomplete or alternative splicing are thus unlikely reasons for the length variation found. Different from VRP1 and VRP2, the product VRP3 was obtained as a unique sequence and exhibited a simple pattern in genomic Southern analysis, as it hybridized to only one detectable fragment.
Expression analysis confirmed that VRP1 is induced during pathogen attack in the resistant cv. ‘Gloire de Montpellier’. In addition, its basal level appeared to be higher than the basic level of the susceptible cv. ‘Riesling’ in Northern hybridizations. In ‘Riesling’ only a very faint increase of VRP1 band intensity could be observed during infection. The difference between resistance and susceptibility in this case hence does not rely on the presence or absence of specific genetic factors, but rather seems to be reflected by different amplitudes and kinetics of gene induction during pathogen defense. How this gene regulation is achieved, awaits further analysis. The fact that the VRP1 sequence shows strong similarity to plant resistance genes revealed it as an interesting candidate gene and encouraged its further analysis. A pathogen-responsive transcript increase could not be confirmed for VRP2 and VRP3, so the investigation of these cDNAs has not yet been pursued.
Using the VRP1 partial sequence from DDRT-PCR the orthologous VRP1 genes were isolated from a BAC-library of the P. viticola resistant cv. ‘Regent’. The positive BAC clone B18G20 was completely sequenced. Three ORFs of this clone are practically identical to the original VRP1 sequences from ‘Gloire de Montpellier’. The identification of three clustered resistance candidate genes on one single BAC clone confirms a small gene family for VRP1. It is possible that additional genes of this family exist in the grapevine genome.
Due to the high similarity to the original VRP1 sequences and considering them as paralogs of a putative resistance gene family, the three ORFs were designated as VRP1-1, VRP1-2 and VRP1-3. Blast analyses and protein motif searches classified these genes as CC-NBS-LRR resistance gene candidates. The NRG1 resistance gene from Nicotiana benthamiana (Peart et al. 2005) shows the highest homology to the VRP1 genes, followed by Arabidopsis thaliana resistance gene ADR1 (Grant et al. 2003). Both are described as CC-NBS-LRR type resistance proteins.
In these domain searches homology was also found between the N-termini of VRP1-1 and VRP1-2 genes and the RPW8 resistance genes (Xiao et al. 2001; Wang et al. 2007) The RPW8 proteins have a transmembrane domain at the N-terminus linked to a CC part (ProSite PS51153). A similar structure is identifiable at the N-termini of VRP1-1 and VRP 1-2, but also in NRG1 and ADR1 at variable stringency. The significance of this finding is not completely clear. If true, it could suggest a kind of chimeric resistance proteins generated by fusion of peptide domains from different classes of proteins involved in plant defense. Pathogen effector specific (gene-for-gene) response that is typically hypothesized for the function of NBS-LRR resistance proteins would be linked to the basal general resistance response attributed to the RPW8 proteins in one single protein. Such “domain shuffling” is thinkable regarding the high abundance of NBS-LRR type proteins identified in the grape genome recently (233 copies, Velasco et al. 2007) and the overall presence of transposable and repetitive elements found (Jaillon et al. 2007). Retrotransposable elements are also surrounding the VRP1 genes on the BAC insert sequence analysed here. Such elements could facilitate illegitimate recombination events giving rise to new proteins joining parts of originally separate genes for diverse functions. The responsiveness of the VRP1 genes to other grape pathogens has to be experimentally tested to determine if they have such a broad level resistance mediating phenotype as observed for RPW8.
Redundancy of genes enables diversification. Investigations have shown that variation at the sequence level, especially in the LRR domain of resistance proteins, can alter the specificity of the recognition of pathogen strains (for review see Hulbert et al. 2001; Xiao 2006). The change of a single amino acid within the LRR domain of the rice blast resistance gene Pi-ta can result in susceptibility to the pathogen (Bryan et al. 2000). Comparisons of the LRRs of the VRP1 genes show some amino acids substitutions, especially when comparing VRP1-1 with the two other genes VRP1-2 and VRP1-3. These substitutions could cause different specificities in the interaction with signal molecules of pathogen(s). The small differences observed between the VRP1 proteins from the resistant ‘Regent’a and the susceptible ‘Pinot noir’ derivative PN40024 may contribute to the contrasting phenotypes of resistance versus susceptibility. Further analyses on the sequence diversity of this gene family will be necessary to draw functional conclusions.
The VRP1 genes were induced upon challenging resistant grapevine with the biotrophic pathogen P. viticola. However they mapped in a genomic region (LG-10) where no stable QTL conferring resistance to pathogens has been detected so far. Assuming that the VRP1 genes would play a major role in the detection of the pathogen by the host and activation of resistance responses, a significant change in the phenotype associated to specific (resistance) alleles may be expected. However, the lack of QTLs in this genomic region is not conclusive. It may mean that the VRP1 gene products require additional gene functions to mediate resistance againt P. viticola, and trans-regulating genes of the VRP1 class could be located within QTL regions providing the phenotypic effects on resistance. This assumption is supported by the fact that the VRP1 homologue NRG1 requires the cofunction of gene N, whose product is a TIR-NBS-LRR type protein (Peart et al. 2005). Another possibility is to assume a more general function of the VRP1 gene products in broad level resistance, as shown for RPW8 (Wang et al. 2007). In this case the known QTLs elaborated for resistance specific to P. viticola and Erysiphe necator (Welter et al. 2007) in ‘Regent’ simply would not cover the same phenotypic trait.
This report is the first to present full sequences of resistance gene analogs from grapevine. The relevance of this class of genes has been recognized in other investigations. The Run1 locus for Erysiphe necator resistance from Muscadinia rotundifolia (a close relative of Vitis sp.) is composed of a NBS-LRR gene family (Barker et al. 2005). RGA markers co-located with genomic regions associated with resistance to different pathogens are known in Vitis sp. (Di Gaspero et al. 2007). However, the way in which these genes act is still unknown. The full genome sequence of grape currently being elaborated should strongly promote these investigations.
References
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Barker CL, Donald T, Pauquet J, Ratnaparkhe MB, Bouquet A, Adam-Blondon A-F, Thomas MR, Dry I (2005) Genetic and physical mapping of the grapevine powdery mildew resistance gene, Run1, using a bacterial artificial chromosome library. Theor Appl Genet 111:370–377
Blaich R (1977) Attempts at artificial mycorrhiza formation in Vitis riparia. Vitis 16:32–37
Bryan GT, Wu KS, Farrall L, Jia Y, Hershey HP, McAdams SA, Faulk KN, Donaldson GK, Tarchini R, Valent B (2000) A single amino acid difference distinguishes resistant and susceptible alleles of the rice blast resistance gene Pi-ta. Plant Cell 12:2033–2046
Chang S, Puryear J, Cairney J (1993) A simple and efficient method for isolating RNA from pine trees. Plant Mol Biol Rep 11:113–116
Church GM, Gilbert W (1984) Genomic sequencing. Proc Natl Acad Sci U S A 81:1991–1995
Dangl JL, Jones JDG (2001) Plant pathogens and integrated defence responses to infection. Nature 411:826–833
Demesure B, Sodzi N, Petit RJ (1995) A set of universal primers for amplification of polymorphic non-coding regions of mitochondrial and chloroplast DNA in plants. Mol Ecol 4:129–131
Di Gaspero G, Cipriani G, Adam-Blondon A-F, Testolin R (2007) Linkage maps of grapevine displaying the chromosomal locations of 420 microsatellite markers and 82 markers for R-gene candidates. Theor Appl Genet 114:1249–1263
Fischer B, Salakhutdinov I, Akkurt M, Eibach R, Edwards KJ, Töpfer R, Zyprian EM (2004) Quantitative trait locus analysis of fungal disease resistance factors on a molecular map of grapevine. Theor Appl Genet 108:505–515
Grant JJ, Chini A, Basu D, Loake GJ (2003) Targeted activation tagging of the Arabidopsis NBS-LRR gene, ADR1, conveys resistance to virulent pathogens. Mol Plant Microbe Interact 16:669–680
Hamiduzzaman MM, Jakab G, Barnavon L, Neuhaus JM, Mauch-Mani B (2005) Beta-Aminobutyric acid-induced resistance against downy mildew in grapevine acts through the potentiation of callose formation and jasmonic acid signaling. Mol Plant Microbe Interact 18:819–829
Hanahan D (1983) Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166:557–580
Hulbert SH, Webb CA, Smith SM, Sun Q (2001) Resistance gene complexes: evolution and utilization. Annu Rev Phytopathol 39:285–312
Jaillon O, Aury J-M, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C, Vezzi A, Legeai F, Hugueney P, Dasilva C, Horner D, Mica E, Jublot D, Poulain J, Bruyere C, Billault A, Segurens B, Gouyvenoux M, Ugarte E, Cattonaro F, Anthouard V, Vico V, DelFabbro C, Alaux M, DiGaspero G, Dumas V, Felice N, Paillard S, Juman I, Moroldo M, Scalabrin S, Canaguier A, LeClainche I, Malacrida G, Durand E, Pesole G, Laucou V, Chatelet P, Merdinoglu D, Delledonne M, Pezotti M, Lecharny A, Scarpelli C, Artiguenave F, Pe ME, Valle G, Morgante M, Caboche M, Adam-Blondon A-F, Weissenbach J, Quetier F, Wincker O (2007) The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature doi: 10.1038/nature06148
Kortekamp A (2001) Charakterisierung der Plasmopara-Resistenz bei Weinreben (Vitis sp). Dissertation, University of Karlsruhe
Kortekamp A (2006) Expression analysis of defence-related genes in grapevine leaves after inoculation with a host and a non-host pathogen. Plant Physiol Biochem 44:58–67
Kortekamp A, Zyprian E (2003) Characterization of Plasmopara-resistance in grapevine using in vitro plants. J Plant Physiol 160:1393–1400
Lee SB, Taylor JW (1990) Isolation of DNA from fungal mycelia and single spores. In Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR protocols: a guide to methods and applications. Academic Press, San Diego, pp 282–287
Liang P, Pardee AB (1992) Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science 257:967–971
Linsmaier EM, Skoog F (1965) Organic growth factor requirements of tobacco tissue cultures. Physiol Plant 18:100–127
Meyers BC, Kozik A, Griego A, Kuang H, Michelmore RW (2003) Genome wide analysis of NBS-LRR-encoding genes in Arabidopsis. Plant Cell 15:809–834
Müller-Roeber B, Pical C (2002) Inositol phospholipid metabolism in Arabidopsis, characterized and putative isoforms of inositol phospholipid kinase and phosphoinositide-specific phopholipase C. Plant Physiol 130:22–46
Pan Q, Wendel J, Fluhr R (2000) Divergent evolution of plant NBS-LRR resistance gene homologues in dicot and cereal genomes. J Mol Evol 50:203–213
Parker JE, Coleman MJ, Szabo V, Frost LN, Schmidt R, van der Biezen EA, Moores T, Dean C, Daniels MJ, Jones JDG (1997) The Arabidopsis downy mildew resistance gene RPP5 shares similarity to the Toll and Interleukin-1 receptors with N and L6. Plant Cell 9:879–894
Peart JR, Mestre P, Lu R, Malcuit I, Baulcombe DC (2005) NRG1, a CC-NB-LRR protein, together with N, a TIR-NB-LRR protein, mediates resistance against tobacco mosaic virus. Curr Biol 15:968–973
Southern EM (1975) Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol 98:503–517
Thomas MR, Scott N (1993) Microsatellite repeats in grapevine reveal DNA polymorphisms when analyzed as sequence-tagged sites (STSs). Theor Appl Genet 86:985–990
Tseng T-C, Tsai T-H, Lue M-Y, H-T Lee (1995) Identification of sucrose-regulated genes in cultured rice cells using mRNA differential display. Gene 161:179–182
Van Ooijen JW, Voorrips RE (2001) JoinMap 3.0, software for the calculation of genetic linkage maps. Plant Research International, Wageningen, The Netherlands
Velasco R, Zharkikh A, Troggio M, Cartwright DA, Cestaro A, Pruss A, Pruss D, Pindo M, FitzGerald LM, Vezulli S, Reid J, Malacarne G, Iliev D, Coppola G, Wardell B, Micheletti D, Macalma T, Facci M, Mitchell JT, Perrazzolli M, Eldredge G, Gatto P, Oyzerski R, Moretto M, Gutin N, Stefanini M, Chen Y, Segala C, Davenport C, Dematte L, Mraz A, Battilana J, Stormo K, Costa F, Tao Q, Si-Ammour A, Harkins T, Lackey A, Perbost C, Taillon B, Stella A, Solovyev V, Fawcett JA, Sterck L, Vandepoele K, Grando SM, Toppo S, Moser C, Lanchbury J, Bogden R, Skolnick M, Sgaramella V, Bhatnagar SK, Fontana P, Gutin A, VandePeer Y, Salamini F, Viola R (2007) A high quality draft consesnus sequence of the genome of a heterozygous grapevine variety. PlosOne 2(12) e:1326 doi: 10.1371/journal.pone.0001326
Wang W, Devoto A, Turner JG, Xiao S (2007) Expression of the membrane-associated resistance protein RPW8 enhances basal defense against biotrophic pathogens. Mol Plant Microbe Interact 20:966–976
Welter LJ, Göktürk-Baydar N, Akkurt M, Maul E, Eibach R, Töpfer R, Zyprian E (2007) Genetic mapping and localisation of quantitative trait loci affecting fungal disease resistance and leaf morphology in grapevine (Vitis vinifera L.). Mol Breed 20:359–374
Wielgoss A, Kortekamp A (2006) Comparison of PR-1 expression in different grapevine culture systems after inoculation with a host and a non-host pathogen. Vitis 45:9–13
Xiao S (2006) Current perspectives on molecular mechanisms of plant disease resistance. In: Teixeira da Silva (ed) Floriculture, ornamental and plant biotechnology: advances and topical issues, 1st edn. Global Science Books, UK, pp 317–333
Xiao S, Ellwood S, Calis O, Patrick E, Li T, Coleman M, Turner JG (2001) Broad-spectrum mildew resistance in Arabidopsis thaliana mediated by RPW8. Science 291:118–120
Zhao S, Ooi SI, Pardee AB (1995) New primer strategy improves precision of differential display. Biotechniques 18:842–850
Acknowledgements
This work was supported by the German Research Foundation (Deutsche Forschungsgemeinschaft, research grants Zy II 3-1 and 3-2) and funds from the German Ministry of Nutrition, Agriculture and Consumers Protection (Bundesministerium für Ernährung, Landwirtschaft und Verbraucherschutz). L.W. was supported through a Ph.D. fellowship provided by the Coordination for the Improvement of Higher Education Personnel (CAPES)-Brasilia/Brazil. We wish to thank Claudia Welsch and Margit Schneider for expert technical assistance and Ludger Hausmann for fruitful discussions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Andreas Kortekamp and Leocir Welter—contributed equally to this work.
Rights and permissions
About this article
Cite this article
Kortekamp, A., Welter, L., Vogt, S. et al. Identification, isolation and characterization of a CC-NBS-LRR candidate disease resistance gene family in grapevine. Mol Breeding 22, 421–432 (2008). https://doi.org/10.1007/s11032-008-9186-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11032-008-9186-2