
Abstract
The carbonic anhydrase II (CAII) deficiency syndrome is a rare autosomal recessive osteopetrosis with renal tubular acidosis (RTA) and cerebral calcifications (MIM259730). CAII deficiency syndrome is caused by mutations in the gene CAII, which encodes the enzyme carbonic anhydrase II. CAII mutations are rarely reported in the Asian population. Here, we described two unrelated CAII deficiency families of Chinese Han origin with clinical and genetic analysis. Altogether, 106 subjects, including 2 probands, 4 unaffected family members from two non-consanguineous Chinese families, and 100 healthy controls were recruited. All seven exons and the exon-intron boundaries of the CAII gene were amplified and directly sequenced. Reverse transcription PCR (RT-PCR) was used to study the effect of splice site mutation. All clinical and biochemical parameters of the probands were collected. Two novel mutations of CAII gene were identified by mutational analysis: A nonsense mutation in exon 4 (c.T381C p.Y127X) in both families; a splice mutation at the splice donor site of intron 3 (c.350+2T>C, IVS3+2T>C) in one family. The splice-site mutation causes exon 3 skipping in patient’s mRNA resulting in an in-frame deletion and a novel premature stop codon. These mutations were predicted to result in a loss of function of CAII. This is the first report of CAII deficiency syndrome in Chinese population. Our findings extent the spectrum of CAII mutations observed in patients with CAII deficiency syndrome.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Osteopetrosis is a rare genetic disorder caused by defects in osteoclast formation and function, which ranges widely in severity. Genetically, osteopetrosis is classified into three types as autosomal recessive osteopetrosis (ARO), autosomal dominant osteopetrosis (ADO) and X-linked inheritance osteopetrosis (XLO), respectively (Del et al. 2008a). For ARO, a range of clinical manifestations has been reported to affect bone and other organ systems. The genes involved in human osteopetrosis are associated with the PH regulation and the formation of osteoclasts, which result in osteoclast-rich osteopetrosis and osteoclast-poor osteopetrosis. Osteoclast-rich osteopetrosis involved genes encode (1) The enzyme carbonic anhydrase II (CAII), which reversibly catalyzes the hydrations of CO2 to HCO3 − (Bolt et al. 2005). (2) The a3 subunit of the vacuolar H+-ATPase of the ruffled border (TCIRG1) (Frattini et al. 2000; Kornak et al. 2000). (3) The ruffled border Cl− conductance (CLCN7) (Cleiren et al. 2001; Frattini et al. 2003). (4) The ostm1 protein (OSTM1) likely associated with the function of the Cl− conductance (Lange et al. 2006; Chalhoub et al. 2003). (5) The plekhm1 protein (PLEKHM1) (Van Wesenbeeck et al. 2007; Del et al. 2008b). Osteoclast-poor osteopetrosis involved genes (1) TNFSF11 (RANKL) and TNFRSF11A (RANK). The TNFSF11 gene encodes the main osteoclast differentiation factor, while its receptor, RANK, is a transmembrane protein expressed on the surface of preosteoclasts and mature osteoclasts (Pangrazio et al. 2012). (2) Sorting Nexin 10 (SNX10), which was involved in the process of osteoclast differentiation and function upon RANKL-stimulation (Pangrazio et al. 2013).
CAII deficiency, referred to as carbonic anhydrase II deficiency syndrome (MIM 259730), is an autosomal recessive osteopetrosis with renal tubular acidosis (RTA) and cerebral calcifications. Patients with CAII deficiency syndrome often present with a combination of proximal and distal renal tubular acidosis, and other clinical manifestations comprise an increased frequency of fractures by adolescence, dental abnormalities, developmental delay, short statures and cranial nerve compression (Whyte 1993). Similar with other types of osteopetrosis due to osteoclast failure, patients with CAII deficiency also present with a classical radiological features, which comprise (a) diffuse sclerosis, affecting the skull, spine, rib, pelvis and appendicular bones, (b) bone remodeling defects at the metaphyses of long bones, such as “erlenmeyer flask appearance”, (c) “bone-within-bone” appearance and “sandwich vertebrae” appearance particularly in the pelvis vertebrae (Stark and Savarirayan 2009). The main biochemical feature of CAII deficiency syndrome is hyperchloremic metabolic acidosis.
CAII has a wide tissue distribution, in bone, kidney (proximal tubules and collecting duct), erythrocytes and glial cells, with osteoclasts being particularly rich in CAII. CAII gene locates on chromosome 8p22.2 (GenBank acc.no., NG_000008.10) and encodes a predicted 260 amino acid protein, carbonic anhydrase II (GenBank acc. no., NP_000058.1). CAII-related mutations have been detected in almost every CAII exon, as well as in several intervening intronic sequences (IVS). IVS mutations are commonly predicted to disrupt proper CAII mRNA splicing (Bolt et al. 2005; Hu et al. 1992; Roth et al. 1992). Up to now, 27 different mutations in the CAII gene have been reported, which were from various ethnic groups including Caucasians, Hispanics, African-Americans, Arab and Asians, and about 65 % of all patients were of Arab origin (Nagai et al. 1997; Hu et al. 1997). However, no mutations have been found in Chinese population previously. In this study, we investigated two Chinese families with RTA and cerebral calcifications. The clinical, laboratory, and radiographic features of these families were described. Two novel mutations of CAII (c.T381C p.Y127X and IVS3+2T>C) were identified by mutational analysis in these two Chinese families. The potential functional consequences of these two mutations were explored.
Materials and methods
Subjects
In this study, two families (FM1 and FM2) with CAII deficiency of Chinese Han origin were recruited. Venous blood samples were collected from unaffected (n = 4) and affected individuals (n = 2). An additional unrelated 100 healthy individuals were recruited as controls. Informed consents and approvals from the Department of Scientific Research at Peking Union Medical College Hospital (PUMCH), the local ethic committee, were obtained before the study.
Clinical assessment
The fasted whole blood samples were placed at room temperature for 30 min and centrifuged at 3000 r/min, 4 °C for 10 min to separate the serum for analysis. Reference ranges were obtained from the central laboratory of PUMCH and were all age/sex/ethnically appropriate. Blood routine examination, biochemistry data and blood gas analysis were performed using routine assays available at the central laboratory of PUMCH. Fasted spot urine was collected, and the urine was analyzed by a Urinary Chemical Analyzer (Clinitek 500, SIEMENS, USA).
Radiographic studies were performed in the Department of Radiology at PUMCH. The X-ray of the skull, spine, pelvis and appendicular bones was performed to detect bone deformities.
The bone mineral density (BMD) of the lumbar spine vertebrae 1–4 (L1-L4) and the left proximal femur, including the femoral neck and total hip were measured using dual energy X-ray absorptiometry densitometer (DXA, GE Lunar, USA) at Department of Radiology at the PUMCH. Height and weight of the subjects were measured using standardized equipment.
Mutational analysis
Genomic DNA were isolated from the venous blood samples using a QIAamp DNA blood extraction kit (Qiagen, Germany) according to the manufacture’s instructions. Total RNA of the FM2-1 (the proband of FM2) was obtained using the QiAamp RNA blood kit (Qiagen, Germany). The 7 exons of the CAII gene were sequenced using 7 pairs of primers to screen for CAII mutations in the two probands. The identified CAII mutations were subsequently investigated in their relatives using the same method. The primers were designed using Gene Runner Primer Analysis Software (Table 1). The exons and exon-intron boundaries of the CAII gene were amplified by polymerase chain reaction (PCR). Taq DNA polymerase (Takara, Japan) and its standard buffer were used in all reactions under the following conditions, initial denaturation at 95 °C for 5 min, followed by 35 cycles at 95 °C for 30 s, 50–60 °C for 30 s, and 72 °C for 50 s. The amplified products were sequenced by an automated sequencer (ABI3730XL) according to the manufacturer’s protocol. Sequence alignment was performed using the Basic Local Alignment Search Tool on the National Center for Biotechnology Information database.
One microgram total RNA was reverse-transcribed using the Goscript™ Reverse Transcription system kit (Promega), to generate complementary DNA (cDNA). The specific primer (P-3F: 5′-AAT GGT CAT GCT TTC AAC GTG-3′/P-4R: 5′-CTT GCC CTT TGT TTT AAT GGA A-3′) was designed as described above. PCR reactions with the primer set were carried out using 2 ul of cDNA. Then PCR products were inserted into the pCRII-TOPO TA vector using the T/A Cloning Kits (Invitrogen). DNA was extracted from 5 randomly selected bacterial clones. The size of the inserted PCR fragment in each clone was determined using PCR with the primer set. Plasmid DNA from bacterial clones containing inserted PCR products of different size was purified and sequenced as described above.
Results
Clinical manifestations
The baseline clinical characteristics and laboratory findings of the two probands and their family members were shown in Tables 2 and 3. The osteopetrosis was assessed by skeletal radiography and DXA examination. Renal tubular acidosis was diagnosed based on their clinical features and laboratory findings. Cerebral calcification was confirmed by brain CT scan.
The proband of family-1(FM1)
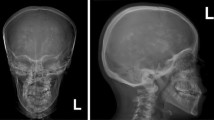
Proband 1 (FM1-1), was the only child of healthy non-consanguineous parents. He went through a full-term normal delivery. The patient couldn’t walk steadily until 1 year and 4 months, and couldn’t speak fluently till 5 years old. He was admitted to our clinic at 6 years of age for the evaluation of failure to thrive and a slight psychomotor retardation. His current height was 106 cm (<−3SD of normal range) and his weight was 18 kg (<−1SD of normal range). Radiological investigation performed and showed a generalized increase in bone density (Fig. 1a, b, c and d), thus leading to the diagnosis of osteopetrosis. The apparent elevation of the bone density was also confirmed by the DXA examination. Oral examination showed his teeth were crowded and misaligned. Laboratory tests showed normal blood cell count, serum calcium and alkaline phosphatase (ALP), but a mild hyperphosphatemia. Blood gas analysis revealed hyperchloremic metabolic acidosis with a PH of 7.232 and a urine pH of 6.5, optimal urinary excretion of acid was not present consistent with a component of distal RTA Brain CT scan showed calcifications in his right frontal lobe (Fig. 1e). Abdominal ultrasound revealed no hepatosplenomegaly. After the treatment of oral citric acid mixture (30 mL tid, 140 g citric acid and 96 g potassium citrate per 1000 mL) and sodium bicarbonate (3.0 g tid) for 11 months, he now has a height of 118 cm (<−1SD of normal range) and a weight of 29 kg (normal range). The latest biochemical examination revealed a serum PH of 7.432 and a urine PH of 8.5.

The clinic characteristics and radiographic signatures of the patients. a–d: Radiographs of skull, femur and appendicular bones of patient 1 showing diffuse increase in bone density. b–c: A typical erlenmeyer flask deformity of distal femur and tibia are indicated by white arrows. d: X-ray of the appendicular bones note metaphyseal lucent bands in the distal ulna and radius (white arrows). e: Brain CT scan of the patient 1 showing intracranial calcification in his right frontal lobe (red arrow). f–h: Radiographs of spine, tibia and pelvis of the patient 2 demonstrates a diffusely increased sclerosis. f: Anteroposterior radiograph of the spine notes the classic vertebral endplate thickening (“sandwich vertebrae” sign) (white arrow). g–h: Fracture lines of tibia and femur are indicated by black arrows. i: Brain CT scan of the patient 2 showing significant intracranial calcification is indicated by red arrow
Both his parents had normal height (father’s height was 187 cm and mother’s height was 165 cm) and without any symptoms of CAII deficiency syndrome.
The proband of family-2 (FM2)
Proband 2 (FM2-1), was the younger of two siblings from healthy non-consanguineous family. She was born at term with normal length and weight. She was diagnosed with CAII deficiency syndrome at 13 years of age, when she was admitted to our hospital for recurrent fractures in 2 years. In the physical examination, a significantly delayed physique and mental development was noted. Her height and weight were 145 cm and 45 kg (both <−2SD of normal range). Physical examination also showed a thoracic deformity including a barrel chest, rachitic rosary and eversion of the costal margin, which were different from the patient 1, suggestive of rickets. Ophtalmological examination showed impaired visual ability. Oral examination showed crowded teeth, a lot of dental caries and normal arcus palatines. Skeletal surveyed by X-ray showed generalized increased bone density with thick cortices and a classic “sandwich appearance” in vertebrae (Fig. 1f). She had histories of frequent fractures since 2012. The first 3 times of fractures happened in 2012 at the right proximal tibia, middle piece of right tibia and right proximal femur with slight damage respectively (Fig. 1g and h), and the last time of fracture happened in 2013 at her right proximal femur again. Laboratory examinations revealed normal levels of serum calcium, phosphate and ALP. Blood gas analysis showed a hyperchloremic metabolic acidosis, with a serum PH of 7.268 and a urine PH of 6.5. Brain CT scan showed intracranial calcification (Fig. 1i). A significantly elevation of the bone mineral density was at lumbar spine and proximal femur found by DXA examination. She started to receive therapy with oral citric acid mixture and sodium bicarbonate (the same dosage with FM1) and was evaluated periodically. Her serum PH elevated to 7.37 after 1 month treatment. The mental development improved when compared with before but still slower than other children of same age.
The height of the patient’s father, mother were 167 and 163 cm, respectively. Unfortunately, the elder sister was unavailable for physical and laboratory examination. None of them had a history of visual loss, frequent fractures, hypokalemia or acidosis.
All the laboratory findings of the two family members were given in Table 3, both of them had normal biochemical parameters.
Genetic analysis
Mutational analysis of the CAII gene in two patients and their parents demonstrated a nonsense mutation in exon 4 (c.T381G, p.Y127X) in FM1 and FM2; a splice mutation at the splice donor site of intron 3 (c.350+2T>C, IVS3+2T>C) in FM2. According to HGMD (http://www.hgmd.org/), the mutations identified were novel.
Patient 1 (FM1-1) was homozygous for a novel nonsense mutation in exon 4 (c.T381G, p.Y127X) causing a premature stop at codon 127. Non-consanguineous parents of patient 1 were heterozygous for this nucleotide change, which were consistent with the mutation in their son (Fig. 2a).

Sequence analysis of human CAII gene and family pedigrees. Patients with CAII deficiency syndrome are depicted by dark symbols, and carriers are depicted by half-dark symbols. a: A novel homozygous nonsense mutation is identified in patient 1, a single base T to G at c.381(Y127X) in exon 4, his father and mother are both asymptomatic heterozygous carrier for this mutation. b: A compound heterozygous mutation was identified in patient 2: a same novel nonsense mutation (Y127X) in one allele from the patient’s father, and a splice mutation (c.350+2T>C) in another allele from the patient’s mother. These two novel mutations were not found in the wild type. The mutational sites in both of the patients are indicated by black arrows. The family member not studied is indicated by “?”
Patient 2 (FM2-1) was compound heterozygous for two novel mutations c.T381G and c.350+2T>C, one of which was also identified in FM1-1. The father of patient 2 was shown to be carrier for the c.T381G mutation, whereas her mother was a carrier for the c.350+2T>C mutation (Fig. 2b).
In order to evaluate the consequences of the c.350+2T>C mutation, we carried out a detailed analysis on mRNA from the patient 2. RNA was firstly reverse-transcribed into cDNA, and amplified by PCR using the special primer set (P-3F/P-4R) (Fig. 3a). As shown in Fig. 3b and c, amplification of patient’s RNA with the special primer set produced two different PCR fragments of 330 and 210 bp, DNA sequencing showed the 330 bp fragment was normal, and the 210 bp fragment corresponded to an mRNA lacking exon 3. This result revealed that this splice-site mutation (c.350+2T>C) caused exon 3 skipping, which in turn resulted in frameshift and was predicted to lead to a truncated protein, causing a novel premature stop codon following a novel 20 amino acid sequence after 77 residues.

Characterization of the effect of IVS3 (c.350+2T>C) point mutation on CAII splicing. a: Schematic partial representation of CAII exon/IVS structure, indicating the position of the primer set (P-3F/P-4R) designed for RT-PCR analysis. b: Clone sequencing of the RT-PCR products from a normal sample reveals the presence of consecutive exons 2 + 3, whereas sequencing of the products from the mutant sample shows the presence of a 120-base deletion, resulting in skipping of exon 3. c: Left, agarose gel showing two different PCR fragments of 330 and 210 amplicons by PCR analysis. P stands for proband, W stands for wild type, M stands for marker. As illustrated on the right, the normal splicing of mRNA encoding the wild-type CAII protein, which contains 260 amino acid residues. The abnormal splicing of mRNA lacking exon 3, and the encoded CAII protein is predicted to bear a premature stop codon following a novel 20 amino acid sequence. The red arrow stands for the splice site
The CAII gene mutations were analyzed in 100 healthy controls, and no identical mutations were detected in the 100 unrelated control samples.
Discussion
Here we described two unrelated Chinese patients, both of whom presented with osteopetrosis, renal tubular acidosis and cerebral calcifications. Other clinical manifestations comprise an increased frequency of fractures by adolescence, short statures, dental abnormalities, cranial nerve compression and developmental delay, consistent with the diagnosis of CAII deficiency syndrome. Genetic analysis showed two novel mutations of CAII: a nonsense mutation in exon 4 (c.T381G, p.Y127X) and a splice mutation at the splice donor site of intron 3 (c.350+2T>C, IVS3+2T>C).
CAII deficiency syndrome is a rare autosomal recessive syndrome which was initially reported by Sly et al. (1983), who demonstrated that only one of the three inherited forms of osteopetrosis is associated with renal tubular acidosis, cerebral calcification. To date, a total of 29 mutations of CAII have been identified, including two novel mutations in the present study. They were distributed in almost all 7 exons and 4 introns, including 6 missense, 4 nonsense, 12 deletions, 1 insertion, 1 deletion-insertion and 5 splice-site mutations. The positions of the identified mutations of CAII exons are shown in Fig. 4, which the most of identified mutations were located in exon 2 and 6, rarely in exon 4. Our discovery of a novel nonsense mutation in exon 4 (Y127X) enriched the database of CAII mutations. The cases in our study are the first report of CAII deficiency syndrome in Chinese.

Sites of known mutations in CAII gene in CAII deficiency syndrome. Boxes in blue & white indicate the 7 exons of CAII gene. The two novel mutations found in this study are labeled in red. The nonsense mutations are indicated by asterisk, the splice mutations are indicated by rhombus
The CAII gene locates in chromosome 8p22.2 and encodes carbonic anhydrase II, which is a cytosolic enzyme consisting of a single polypeptide chain of 260 amino acid residues (Halder and Taraphder 2013). CAII is one of the most efficient enzyme and it reversibly catalyzes the hydrations of CO2 to HCO3 −, where it acts on such important processes as the bicarbonate reabsorption (kidney) and the bone resorption (Tashian 1989). Thus deficiency of CAII results in a syndrome of osteopetrosis, renal tubular acidosis and cerebral calcification. The structure of CAII includes helix bundles, sheets, the substrate binding region, the zinc ion binding site and the active sites (Hakansson et al. 1992). Eriksson et al. (1988) constructed a three-dimensional structure of human CAII by the homology modeling technique and suggested the roles of specific amino residues in protein fold: zinc ion binding site (94H, 96H, 119H) and the active site (64H, 67N, 107H, 127Y) (Fig. 5). Subsequently, Halder and Taraphder (2013) presented molecular modeling of the structure and possible proton transfer pathways from the surface of the protein to the zinc-bound water molecule in the active sites of human carbonic anhydrase II, and showed that the presence or absence of the complete proton transfer pathway depends crucially on the water structure at the active sites. Thus, the nonsenses mutation (Y127X) might be associated with a severe clinical consequence since the mutated protein was located at the active site, which would result in absence of CAII activity due to absence of the complete proton transfer pathway.

The three-dimensional structure of wild-type and mutated CAII protein. a: The three-dimensional structure of human CAII protein constructed by Eriksson et al. (1988), in which the zinc ion binding site and its three His residues (94His, 96His, 119His) have been labeled blue. The active sites are labeled violet and hot pink, which contain the amino acid residues (64His, 67Asn, 107His, 127Lys). The mutated amino acid 107His which is labeled hot pink has been demonstrated to retain residual activity in the active site of CAII. The protein backbone is represented as green ribbon. b: The three-dimensional structure of mutated CAII protein (c.T381C p.Y127X). The nonsence mutation causes a premature stop at codon 127
Clinical heterogeneity has been reported in CAII deficiency syndrome patients. Soda et al. (1995) first reported a nonsense mutation in exon 2 (Y40X) in a Japanese patient who exhibited severe mental retardation except for the other features of CAII deficiency syndrome. Subsequently, Shah et al. (2004) reported another two nonsense mutations in a Turkish patient (Q28X) and Italian patient (W97X). As the Japanese patient, the Turkish patient also presented with a classic CAII deficiency syndrome and severe mental retardation, whereas the Italian patient exhibited only renal tubular acidosis and mild osteopetrosis without intracranial calcification and mental retardation. It is supposed that heterogeneity is related to complete absence of CAII activity or partial absence of CAII or other genes of CA isozymes, such as CAI. In the present study, patient 1 only presented with a classic CAII deficiency syndrome: osteopetrosis with RTA and cerebral calcifications. Compared with patient 1, patient 2 had much more severe clinical phenotype, except the classic CAII deficiency syndrome, who also exhibited cranial nerve compression with visual impairment, frequently fractures, severe mental retardation and rickets. Patient 1 was homozygous for a nonsense mutation (Y127X), whereas, patient 2 was compound heterozygous for a nonsense mutation (Y127X) in one allele in exon 4 as patient 1, and a novel splice mutation (350+2T>C) in another allele. The splice mutation caused a decrease in splicing efficiency with skipping of exon3, resulted in frameshift and was predicted to lead to premature stop at codon 97, which leads to the production of a truncated protein which lacks the 163 amino acid sequence at the C-terminus. It was supposed that the splice mutation leading to shorter truncated products might be related to the severe clinical phenotype of patient 2. Four CAII splice site mutations have been reported in the literatures (Hu et al. 1992, 1997; Roth et al. 1992; Nampoothiri and Anikster 2009), but none of these have been reported in Chinese population.
There are some limitations in our study. Firstly, the number of subjects is not big enough to reveal the phenotype-genotype correlations. Secondly, we did not conduct a functional analysis of the mutant CAII protein of our patient. Further investigations are needed to identify the correlation between phenotype and genotype, also need to provide considerable insight into the structure function relationship in CAII.
In conclusion, we enriched the spectrum of mutations in CAII by identifying two novel mutations in two Chinese families with CAII deficiency syndrome. Careful clinical observation in combination with molecular genetic analysis would enrich the current understanding of CAII deficiency syndrome considerably. We supposed that truncation mutation and nonsense mutation can lead to shorter truncated products, which might be related to the severe clinical phenotypes in the patient. Future analysis of more cases is still needed to clarify the distribution and frequency of CAII gene mutations and the phenotype-genotype relationship in Chinese patients with CAII deficiency syndrome.
References
Bolt RJ, Wennink JM, Verbeke JI, Shah GN, Sly WS, Bokenkamp A (2005) Carbonic anhydrase type ii deficiency. Am J Kidney Dis 46(A50):e71–e73
Chalhoub N, Benachenhou N, Rajapurohitam V, Pata M, Ferron M, Frattini A, Villa A, Vacher J (2003) Grey-lethal mutation induces severe malignant autosomal recessive osteopetrosis in mouse and human. Nat Med 9:399–406
Cleiren E, Benichou O, Van Hul E, Gram J, Bollerslev J, Singer FR, Beaverson K, Aledo A, Whyte MP, Yoneyama T, DeVernejoul MC, Van Hul W (2001) Albers-schonberg disease (autosomal dominant osteopetrosis, type ii) results from mutations in the clcn7 chloride channel gene. Hum Mol Genet 10:2861–2867
Del FA, Cappariello A, Teti A (2008a) Genetics, pathogenesis and complications of osteopetrosis. Bone 42:19–29
Del FA, Fornari R, Van Wesenbeeck L, de Freitas F, Timmermans JP, Peruzzi B, Cappariello A, Rucci N, Spera G, Helfrich MH, Van Hul W, Migliaccio S, Teti A (2008b) A new heterozygous mutation (r714c) of the osteopetrosis gene, pleckstrin homolog domain containing family m (with run domain) member 1 (plekhm1), impairs vesicular acidification and increases tracp secretion in osteoclasts. J Bone Miner Res 23:380–391
Eriksson AE, Jones TA, Liljas A (1988) Refined structure of human carbonic anhydrase ii at 2.0 a resolution. Proteins 4:274–282
Frattini A, Orchard PJ, Sobacchi C, Giliani S, Abinun M, Mattsson JP, Keeling DJ, Andersson AK, Wallbrandt P, Zecca L, Notarangelo LD, Vezzoni P, Villa A (2000) Defects in tcirg1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nat Genet 25:343–346
Frattini A, Pangrazio A, Susani L, Sobacchi C, Mirolo M (2003) Chloride channel clcn7 mutations are responsible for severe recessive, dominant, and intermediate osteopetrosis. J Bone Miner Res 18:1740–1747
Hakansson K, Carlsson M, Svensson LA, Liljas A (1992) Structure of native and apo carbonic anhydrase ii and structure of some of its anion-ligand complexes. J Mol Biol 227:1192–1204
Halder P, Taraphder S (2013) Modeling the structure and proton transfer pathways of the mutant his-107-tyr of human carbonic anhydrase ii. J Mol Model 19:289–298
Hu PY, Roth DE, Skaggs LA, Venta PJ, Tashian RE, Guibaud P, Sly WS (1992) A splice junction mutation in intron 2 of the carbonic anhydrase ii gene of osteopetrosis patients from Arabic countries. Hum Mutat 1:288–292
Hu PY, Lim EJ, Ciccolella J, Strisciuglio P, Sly WS (1997) Seven novel mutations in carbonic anhydrase ii deficiency syndrome identified by sscp and direct sequencing analysis. Hum Mutat 9:383–387
Kornak U, Schulz A, Friedrich W, Uhlhaas S, Kremens B, Voit T, Hasan C, Bode U, Jentsch TJ, Kubisch C (2000) Mutations in the a3 subunit of the vacuolar h(+)-atpase cause infantile malignant osteopetrosis. Hum Mol Genet 9:2059–2063
Lange PF, Wartosch L, Jentsch TJ, Fuhrmann JC (2006) Clc-7 requires ostm1 as a beta-subunit to support bone resorption and lysosomal function. Nature 440:220–223
Nagai R, Kooh SW, Balfe JW, Fenton T, Halperin ML (1997) Renal tubular acidosis and osteopetrosis with carbonic anhydrase ii deficiency: pathogenesis of impaired acidification. Pediatr Nephrol 11:633–636
Nampoothiri S, Anikster Y (2009) Carbonic anhydrase ii deficiency a novel mutation. Indian Pediatr 46:532–534
Pangrazio A, Cassani B, Guerrini MM, Crockett JC, Marrella V (2012) Rank-dependent autosomal recessive osteopetrosis: characterization of five new cases with novel mutations. J Bone Miner Res 27:342–351
Pangrazio A, Fasth A, Sbardellati A, Orchard PJ, Kasow KA (2013) Snx10 mutations define a subgroup of human autosomal recessive osteopetrosis with variable clinical severity. J Bone Miner Res 28:1041–1049
Roth DE, Venta PJ, Tashian RE, Sly WS (1992) Molecular basis of human carbonic anhydrase ii deficiency. Proc Natl Acad Sci U S A 89:1804–1808
Shah GN, Bonapace G, Hu PY, Strisciuglio P, Sly WS (2004) Carbonic anhydrase ii deficiency syndrome (osteopetrosis with renal tubular acidosis and brain calcification): novel mutations in ca2 identified by direct sequencing expand the opportunity for genotype-phenotype correlation. Hum Mutat 24:272
Sly WS, Hewett-Emmett D, Whyte MP, Yu YS, Tashian RE (1983) Carbonic anhydrase ii deficiency identified as the primary defect in the autosomal recessive syndrome of osteopetrosis with renal tubular acidosis and cerebral calcification. Proc Natl Acad Sci U S A 80:2752–2756
Soda H, Yukizane S, Yoshida I, Aramaki S, Kato H (1995) Carbonic anhydrase ii deficiency in a Japanese patient produced by a nonsense mutation (tat–>tag) at tyr-40 in exon 2, (y40x). Hum Mutat 5:348–350
Stark Z, Savarirayan R (2009) Osteopetrosis. Orphanet J Rare Dis 4:5
Tashian RE (1989) The carbonic anhydrases: widening perspectives on their evolution, expression and function. Bioessays 10:186–192
Van Wesenbeeck L, Odgren PR, Coxon FP, Frattini A, Moens P, Perdu B, MacKay CA, Van Hul E, Timmermans JP, Vanhoenacker F, Jacobs R, Peruzzi B, Teti A, Helfrich MH, Rogers MJ, Villa A, Van Hul W (2007) Involvement of plekhm1 in osteoclastic vesicular transport and osteopetrosis in incisors absent rats and humans. J Clin Invest 117:919–930
Whyte MP (1993) Carbonic anhydrase ii deficiency. Clin Orthop Relat Res 52–63
Acknowledgments
We appreciate our patients and their families for their participating in this study.
This study was supported by a grant from The Ministry of Science and Technology of the People’s Republic of China (National Science and Technology Major Projects for “Major New Drugs Innovation and Development” 2008ZX09312-016). National Natural Science Foundation of China (No. 81070687, 81470188 and 81170805), Beijing Natural Science Foundation (No. 7121012) and Scientific Research Foundation of Beijing Medical Development (No. 2007–3029). National Key Program of Clinical Science (WBYZ2011-873)
All listed authors have each made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; participated in drafting the manuscript or revising it critically for content; and have approved the final version of the submitted manuscript.
Dr. Qian-qian Pang and Prof. Wei-Bo Xia accept responsibility for the integrity of the data analysis.
Conflict of interest
All authors have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Pang, Q., Qi, X., Jiang, Y. et al. Two novel CAII mutations causing carbonic anhydrase II deficiency syndrome in two unrelated Chinese families. Metab Brain Dis 30, 989–997 (2015). https://doi.org/10.1007/s11011-015-9660-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-015-9660-6