
Abstract
The interleukin-6 (IL-6) is a pleiotropic cytokine that plays a key role in interaction between immune and nervous system. Although IL-6 has neurotrophic properties and beneficial effects in the CNS, its overexpression is generally detrimental, adding to the pathophysiology associated with CNS disorders. The source of the increase in peripheral IL-6 remains to be established and varies among different pathologies, but has been found to be associated with cognitive dysfunction in several pathologies. This comprehensive review provides an update summary of the studies performed in humans concerning the role of central and peripheral IL-6 in cognitive dysfunction in dementias and in other systemic diseases accompained by cognitive dysfuction such as cardiovascular, liver disease, Behçet’s disease and systemic lupus erythematosus. Further research is needed to correlate specific deficits in IL-6 and its receptors in pathologies characterized by cognitive dysfunction and to understand how systemic IL-6 affects high cerebral function in order to open new directions in pharmacological treatments that modulate IL-6 signalling.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Interleukin-6 (IL-6) belongs to the neuropoietic cytokines family which members have a molecular weight of 21 kDa in their non-glycosylated form (May et al. 1991). The IL6 gene contains four introns and five exons and it is located at chromosome 7p21 in the human genome. The cytokine is synthesized by a variety of cells including monocytes, fibroblasts, endothelial cells and various cell types of central nervous system (CNS), and its concentration is modulated by numerous factors including proinflammatory agents, viral and bacterial pathogens, neurotransmitters, and second messengers (Gruol and Nelson Gruol and Nelson 1997). The complex regulation of IL-6 gene expression is provided by different binding sites for transcription factors including those for nuclear factor KB (NF-KB), nuclear factor-IL-6 (NF-IL-6), and activator protein-i, and two glucocorticoidresponsive elements (GRE 1 and GRE2) (Dendorfer et al. 1994). Under physiological conditions, IL-6 levels in the CNS are low, while its levels increase during brain injury, inflammation, and disease. Another cause that leads to an increase in brain IL-6 concentration in the brain is the net influx from to blood into brain parenchyma leading to an eventual increase in central production of inflammatory cytokine (Di Santo et al. 1996).
In this review we present an update summary of the role of IL-6 in cognitive impairment in dementias and systemic disease that manifest with cognitive impairment.
In particular we structured the review in the following sections:
-
Distribution of IL-6 and its receptors in the brain
-
The roles of IL-6 in the brain
-
Alterations of central and peripheral IL-6 concentration in Alzheimer’s disease, vascular dementia, Levy body dementia.
-
The relationship between IL-6 concentration in blood and cognitive impairment in pathologies accompanied by an increase production of the cytokine (cardiovascular, liver disease, Behçet’s disease and systemic lupus erythematosus).
-
The mechanisms by which IL-6 may induce cognitive impairment.
-
Distribution of IL-6 and its receptors in central nervous system
IL-6 activates a cell surface signalling assembly comprised of two membrane proteins, the IL6Rα is the ligand binding α-subunit receptors and gp130, the signal transducing β- subunit. The mature form of IL6-Rα is a glycosylated 80 kDa derived from 50 kDa precursor protein (Varghese et al. 2002. Firstly IL-6 binds to IL- 6Rα and then presented to gp130 in the proper geometry to facilitate a cooperative transition into the high-affinity, signaling-competent hexamer (Gruol and Nelson 1997; Boulanger et al. 20033). The IL6-Rα also exists in a soluble form (Peters et al. 1998). In contrast to other soluble cytokine receptors, the complex formed by IL-6 with its soluble receptor is able to interact with gp130 on target cells, thereby triggering the IL-6 signal transduction pathway on cells, which do not express the IL-6R, and which are unresponsive to IL-6 (Fig. 1). This process has been called trans-signaling IL-6 and it can take place on all cells of the body since all cells express gp130 (Rose-John 2012 for review). Distinct cytokines that display overlapping biological activities possess a ligand-binding α-chain and signal-α-transducing β-chain. IL-6 belongs to family of citokines (with IL-11, oncostatin M, leukemia inhibitory factor, ciliary neurotrophic factor and cardiotrophin-1 (CT-1), cardiotrophin-like cytokine (CLC), neuropoietin (NPN), IL-27 and IL-31) that bind to the β-chain of gp130 receptor (Boulanger et al. 2003). Stimulation of gp130 receptors leads to intracellular activation of Src and Janus tyrosine kinases, STAT transcription factors and Ras-mediated signalling (Heinrich et al. 1998; Chow et al. Chow et al. 2001). In the periphery, IL-6 stimulates several types of leukocytes, the production of acute phase proteins in the liver and activation of acute-phase plasma proteins genes (Gauldie et al. 1987). IL-6 plays a key role in the differentiation of B lymphocytes (Kopf et al. 1994) and protein breakdown in muscle (Goodman 1994).

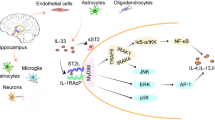
Schematic representation of IL-6, its receptors and the signal transducer gp130. (A) IL-6 first binds to the membrane-bound IL-6R. This complex associates with the protein gp130 thereby inducing JAK kinase activation, which in turn induces the activation of STAT family proteins. The activated STAT proteins translocate into the nucleus and activate intracellular signalling. (B) the complex formed by IL-6 with its soluble receptor (sIL-6R) is able to interact with gp130 on target cells, thereby triggering the IL-6 signal transduction pathway on cells, which do not express the IL-6R (trans-signalling pathway)
Expression of IL-6 and its receptors in the brain
Under physiological conditions IL-6 mRNA is almost undetectable in the rat brain, but its transcription cab be increased after inflammatory signal such as administration of bacterial lipopolysaccharide (LPS). The increase in IL-6 mRNA occurs in the choroid plexus and the sensorial circumventricular organs (CVOs) such organum vasculosum lamaniae terminalis, subfornical organ, median eminence and area postrema (Vallières and Rivest 1997). These brain areas are located outside the blood brain barrier (BBB) thus they can transmit the information coming from the periphery to specific brain areas isolated by the BBB. Hampel et al. (2005)) analyzed the expression of IL-6 in five regions of the human brain (frontal-, temporal-, parietal-, occipital cortex and cerebellum) and found that it is extensively distributed in white matter of these brain areas. IL-6 is expressed in many cell type of CNS, i.e. astroglia, microglia, neurons and endotelial cells (Gadient and Otten 1997. Oligodendrocytes seem do not express IL-6 receptor mRNA (Sawada et al. 1995). In contrast to the faint IL-6 mRNA, the expression of IL-6 R mRNA is stronger and it’s heterogeneously distributed in the rat brain. The strongest expression is observed in the pyramidal cells of CA1-CA4 regions of the hippocampus, the granular cell layer of the dentate gyrus and the cerebellum. In the cerebral cortex, IL-6 R mRNA is abundant in septohippocampal nucleus, claustrum and layer 2 of pyriform cortex. Lower staining is observed in other cortical regions, bed nucleus of stria terminalis, central nucleus of amygdale ependymal lining cells of the ventricular system, the CVOS and the vascular organ of the lamina terminalis. White matter regions also show IL-6 R mRNA expression (Gadient and Otten 1993; Schöbitz et al. 1993; Gadient and Otten 1994a). LPS stimulates the expression of IL-6 receptor mRNA in CVOs, medial preoptic area, bed nucleos of stria terminalis, central nucleus of amygdala, hippocampus, paraventricular nucleus of the hypothalamus, pyriform and septohippocampal cortex, claustrum and in brain blood vessels (Vallières and Rivest 1997). IL-1 β stimulates the expression of IL-6 R mRNA in blood vessels although less intensely than LPS. Basal expression of IL-6 and IL-6 receptors mRNA has also been observed by using oligonucleotide probes in several brain areas i.e. the pyriform cortex, pyramidal and granular neurons of the hippocampus, hypothalamus, habenula and granular cells of the cerebellum and white matter (Schöbitz et al. 1993). The fact that expression of IL-6 and its receptors is detectable under physiological conditions suggests that it may regulate physiological functions of brain cells.
Expression of the signal transducer gp130
gp130 mRNA is present in high quantities and is heterogeneously distributed in rat brains. Higher gp130 expression compared to IL-6 receptors is not surprising since gp130 serves as signal transducer for IL-6 and other cytokines as mentioned above. In contrast to IL-6 receptor mRNA, gp130 mRNA is absent in white matter regions such as the anterior commissure, corpus callosum, fornix, and optic tract (Vallières and Rivest 1997). gp130 mRNA expression is increased after lipopolysaccharide (LPS) infusion, but remains unchanged following IL-1 β infusion into the organum vasculosum laminae terminalis and the endothelium of brain capillaries (Vallières and Rivest 1997). Membrane-bound gp130 localises to astroglia, microglia, neurons and endothelial cells in human controls, and its expression is higher compared to IL-6 and IL-6R (Hampel et al. 2005). Membrane-bound gp130 consistently shows strong expression in all brain regions, with no prominent differences between frontal-, temporal-, parietal-, occipital cortex, and cerebellum tissues (Hampel et al. 2005).
Roles of IL-6 in the brain
IL-6 is generally viewed as a destructive, proinflammatory cytokine which induces inflammatory acute-phase protein expression; these proteins act as major pyrogens increasing vascular permeability, lymphocyte activation, and antibody synthesis. Although IL-6 overexpression is generally detrimental and can add to the pathology of several CNS disorders, there is evidence that IL-6 may also have anti-inflammatory and immunosuppressive properties, regulates neuronal survival and function (Guzmán and Hallal-Calleros 2010) at low levels. In the CNS, IL-6 can also act as an indirect immunosuppressant because it potently stimulates the pituitary-adrenal axis (Mastorakos and Ilias 2006; Girotti et al. 2012) eliciting the release of adrenocorticotropic hormones that, in turn, increase the synthesis of glucocorticoids by the adrenal gland. IL-6 also inhibits interferon-g (IFN-g), IL-1 β, and lipopolysaccharide (LPS)-induced synthesis of TNF-α protein in glial cells at the post-transcriptional and/or post-translational level (Shrikant et al. 1995). IL-6 promotes in vitro survival of acetylcholinesterase-positive neurons from embryonic rat spinal cord (Kushima and Hatanaka 1992) and catecholaminergic neurons from fetal and postnatal rat midbrains (Kushima et al. 1992). IL-6 also protects cultured hippocampal neurons against glutamate-induced death, excitoxicity induced by the glutamate receptor agonist NMDA in rat striatal cholinergic neurons in vivo (Toulmond et al. 1992), MPP+ neurotoxicity (Akaneya et al. 1995), ischemia and nerve injury (Hirohata 1996). Moreover, IL-6 may also act on oligodendrocyte survival in the nervous system (Barres et al. 1993) as well as regulating food intake and sleep (Kent et al. 1992; Wallenius et al. 2002).
It has also been suggested that IL-6 may be involved in repair mechanisms following traumatic brain injury by increasing nerve growth factor secretion by astrocytes (Kossmann et al. 1996). Evidence has also been found that IL-6 acts as a neuroprotector in diabetic neuropathy (Andriambeloson et al. 2006). The stability of this IL-6/sIL-6R/gp130 complex determines the balance between these three proteins and thus determines if an IL-6-specific signal is initiated, thus switching IL-6-mediated effects from neuroprotection to neurotoxicity.
Among its central functions, IL-6 increases body temperature, disrupts the blood–brain barrier (BBB) and stimulates gliosis (Schöbitz et al. 1994). Like the resident immunocompetent cells such as astrocytes or microglia, neurons can also release IL-6 both in vivo and in vitro. Although some reports indicate that neurons express IL-6 under normal physiological conditions (Schöbitz et al. 1993; Gadient and Otten 1994b; März et al. 1997), this expression is profoundly increased in cerebral ischemia and after axotomy (Murphy et al. 1995; Suzuki et al. 1999). IL-6 expression in neurons contributes to glial cell activation (Klein et al. 1997), protects neighbouring neurons (Hama et al. 1991; Yamada and Hatanaka 1994), and supports the regeneration of peripheral nerves after lesion (Zhong et al. 1999). However, chronic neuronal exposure to IL-6 increases Ca2+ influx in response to NMDA (N-methyl-D-aspartate) which causes cell death (Qiu et al. 1998).
Role of IL-6 in cognitive impairment
Cognitive symptoms related to IL-6 include the core clinical signs of dementia, and other systemic pathologies (liver cirrhosis, cardiovascular diseases, etc.) that can also present cognitive impairment. In the following sections we separately review these data because the conclusions that can be drawn from differ between pathologies.
IL-6 in Alzheimer’s disease
Alzheimer’s disease (AD) is a leading cause of dementia in the elderly population. Its principal pathological hallmarks are the deposition of extracellular Aβ plaques, formation of intracellular neurofibrillary tangles, and features of general brain inflammation: activation of microglia, astrocytosis, and production of several proinflammatory mediators, including IL-6. Bauer et al. (1992) first reported increased IL-6 expression in AD patient brain tissues at autopsy, and since then many studies have analysed the concentration of IL-6 in AD patient brain, cerebrospinal fluid (CSF), and blood samples (See Table 1).
Hampel et al. (2005) found a consistent increase in IL-6 and IL-6R expression in AD patient brain sections, although membrane-bound gp130 is strongly and uniformly expressed in both control and AD individuals. In particular, IL-6 expression is increased in the parietal cortex and decreased in occipital and temporal cortices compared to controls, whereas expression of IL-6R is strongly increased in frontal and occipital cortices and decreased in temporal cortex and the cerebellum compared to controls (Hampel et al. 2005). However, the decreased levels of IL-6 in the temporal cortex reported by Hampel et al. (2005) do not agree with those found by Wood et al. (1993) who reported increased IL-6 concentrations in AD temporal cortex homogenates compared to controls. Moreover, (Hull et al. (1996a; 1996b) , did not find IL-6 expression in the brains of elderly control subjects but did find increased levels in AD patient plaques. These discrepancies may be attributable to methodological differences, since ELISA assays on brain tissue homogenates are very limited in their reliability and reproducibility.
Also worth considering is that patients with late-onset AD have significantly higher IL-6 levels compared to those with early-onset AD (Jellinger 2009). Huell et al. (1995) investigated the appearance of IL-6 immunoreactivity in AD plaques according to the stage of plaque formation and showed that IL-6 appeared to be linked to early plaque-formation stages, suggesting that it might be a factor related to the early histopathological changes occurring in neurodegenerative processes. Some reports also show that levels of CSF IL-6 are not altered in patients with AD (Tarkowski et al. 2003; Wada-Isoe et al. 2004), which directly contrasts with the findings of other studies (Jia et al. 2005; Jellinger 2009). However, careful analysis of the clinical history of these AD patients suggests that patients with high IL-6 levels had a relatively short disease history, suggesting a relationship between CSF IL-6 levels with a longer duration and higher degree of dementia. Several studies have evaluated the concentration of IL-6 in AD patients’ blood, and although several reports described an elevated peripheral blood IL-6 concentration in AD (Licastro et al. 2000; Akiyama et al. 2000; Engelhart et al. 2004) there are other conflicting results (see Table 1).
Interestingly, activation of the CNS inflammatory response in rats by intracerebroventricular injection of IL-1b results in an increased serum IL-6 concentration (De Simoni et al. 1993). Moreover, the same experiment in primates resulted in increased IL-6 concentrations in both CSF and blood (Xiao et al. 1999). Thus, it seems that the CNS is not only able to produce IL-6, but can also contribute to the peripheral cytokine pool. Indeed, many investigators support the hypothesis that systemic IL-6 production outside the CNS can affect brain function and cognition in inflammatory disorders, but this mechanism of pathogenesis is difficult to establish empirically, and so far has always been induced by other cytokines (e.g. IL-1) or by LPS administration. Shalit et al. (1994) demonstrated that phytohaemagglutinin (PHA)-stimulated IL-6 release in mononuclear cells is increased in AD patients, although this increase was not apparent in patients with late onset AD (Sala et al. 2003; Zuliani et al. 2007b). IL-6 secretion is also decreased in severely demented patients versus controls, suggesting that it might negatively correlate with the severity of dementia (Sala et al. 2003), which also agrees with in vitro study findings suggesting that IL-6 acts as a neuroprotective molecule (Tarkowski et al. 2003). However, normal serum IL-6 levels were found in the early stages of late-onset sporadic AD (van Duijn et al. 1990; Kalman et al. 2009) suggesting AD subtype might play a role in the discrepancies observed between studies. Confounding these findings, increases in IL-6 serum levels may relate to several factors, including aging (Kalman et al. 1997; Ershler and Keller 2000) or the presence of latent infectious, inflammatory, or malignant diseases in the late stages of the dementia.
Nevertheless, normal serum and CSF IL-6 levels in the mild-moderate stage of AD do not exclude local elevation in the CNS (Berkenbosch et al. 1992; Nybo et al. 2002). Indeed, intrathecal inflammation has been reported to precede the development of AD in patients who initially presented with mild cognitive impairment (Tarkowski et al. 2003); this could be explained by increased BBB permeability, allowing IL-6 to enter the brain more easily, and also explaining why IL-6 levels are increased in the CSF but not in blood in the late stages of AD. The positive correlation between blood serum IL-6 levels and the severity of dementia could indicate that this cytokine might be a stage marker rather than a state marker in AD. Although many studies have failed to show this correlation, it has been proposed that genetic polymorphisms may play a role in this variability (Licastro et al. 2003).
IL-6 in Lewy body dementia
Dementia with Lewy bodies (DLB) is the second most common form of dementia following Alzheimer’s disease (AD), which account for about 20% and 60% of dementias respectively. DLB tends to affect males more than females, with the typical age of onset between 50 and 80, and the illness usually lasting about 6 years before death (Cercy and Bylsma 1997). DLB generally shows a more rapid progression than AD, which may be due to the neuroleptic medication sometimes used, as it is associated with an increase in morbidity and mortality rates. Clinical diagnosis of DLB includes the presence of three core features: fluctuating alertness/cognition, recurrent fully-formed visual hallucinations, and spontaneous parkinsonism (McKeith et al. 1996). The defining neuropathological features of DLB are the presence of Lewy bodies, Lewy neurites, and spongiform encephalopathy in the amygdala, entorrhinal cortex and temporal gyrus, paralimbic and neocortical regions, and substantia nigra (McKeith et al. 1996). Lewy bodies are rounded intracytoplasmic neuronal inclusions and Lewy neurites are diffuse and filamentous inclusions. Patients with DLB exhibit a diffuse plaque-only pathology with rare neocortical neurofibrillary tangles as opposed to the widespread neurofibrillary-tau tangles observed in AD patients. Increased mRNA IL-6 expression, as measured by RT-PCR, has been reported in the hippocampus, putamen, and cingulated cortex in the brains of patients who died with DLB compared to undetectable expression in control subjects (Imamura et al. 2004). The increase in IL-6 and other pro-inflammatory cytokines such IL-1 β and TNF-α is likely due to the increased expression of microglia cells in these brain structures (Imamura et al. 2004; Sawada et al. 2006). Interestingly, identical changes were observed in the same brain areas of patients with Parkinson’s disease, suggesting a link to the neurodegenerative processes occurring in these neurodegenerative diseases (Imamura et al. 2004; Sawada et al. 2006). CSF IL-6 concentration is not significantly increased in DLB patients, indicating a selective, rather than general, increase in specific brain areas (Gómez-Tortosa et al. 2003). To our knowledge, no data have been published regarding changes in blood IL-6 concentration in DLB patients.
IL-6 in vascular dementia
Vascular dementia (VD) encompasses many clinical syndromes because it can result from a variety of pathogenic mechanisms (Amos 2002). VD is defined as the loss of cognitive function resulting from schemic-hypoxic, or haemorrhagic brain tissue lesions due to cardiovascular disease and cardiovascular pathological changes (Baskys and Cheng 2012). In contrast to the highly variable results obtained in AD, there is a consensus in the literature about the increase of IL-6 concentration in plasma and CSF in patients with VD, suggesting that inflammatory mechanisms may be involved in the development of cognitive impairment in some patients with cerebrovascular disease (Wada-Isoe et al. 2004; Jia et al. 2005; Zuliani et al. 2007b). The increase in IL-6 concentration in plasma and CSF in VD patients is higher than that observed in AD patients (Wada-Isoe et al. 2004; Jia et al. 2005; Zuliani et al. 2007a; Zuliani et al. 2007b). In addition, functional status (assessed on the Barthel score) is inversely associated with IL-6 blood plasma concentration (Zuliani et al. 2007a; Zuliani et al. 2007b), although the mechanisms linking IL-6 levels and functional status have not yet been identified. High levels of IL-6 have been associated with a number of diseases (including atherosclerosis, coronoray hearth disease, stroke, hypertension, congestive heart failure, and diabetes) which are more common in VD and are linked to disability (Fried and Guralnik 1997; Krabbe et al. 2004). Moerover, high IL-6 promotes anaemia, fatigue, muscle wasting, and a reduction in muscle strength (Visser et al. 2002; Cesari et al. 2004) which can cause a decline in the functional status of patients. The association between IL-6 levels and Mini-Mental State Examination (MMSE) score is not significant in patients with VD, therefore suggesting that elevated peripheral IL-6 does not accurately reflect the degree of brain damage and/or functional impairment caused by this cytokine.
The reason for increased CSF IL-6 in VD patients could be due to reactive inflammation in the CNS observed in some cardiovascular disease (Egashira 2003) and/or to brain ischemia and hypoxia that in turn, leads to degeneration of nerve fibers. When this damage occurs, glial cells react to clear damage fibers and produce reactive inflammation leading to overproduction of inflammatory cytokines included IL-6 (Stys and Jiang 2002). Finally, a genetic association of IL-6 polymorphism with multi-infarct dementia has been demonstrated (Pola et al. 2002).
IL-6 and cognitive impairment in patients with cardiovascular disease
Cognitive impairment after hearth stroke is characterized by deficits in global cognition, as well as domain-specific impairments involving executive function, language, visuospatial ability, and/or memory in a third of patients (Tatemichi et al. 1994). Interestingly, peripheral IL-6 concentration significantly correlates with MMSE in post-cardioinfarction patients (Rothenburg et al. 2010). Cognitive decline after coronary artery bypass grafting (CABG) is also recognized and well documented (Hammeke and Hastings 1988; Borowicz et al. 1996). Additionally, microembolism, brain hypoperfusion, non-pulsatile flow, and inflammatory responses during cardiopulmonary bypass (CPB), also result in a similar cognitive impairment (Henriksen et al. 1983; Pugsley et al. 1994), however there is little agreement on the mechanisms which underlie it. The incidence of cognitive decline after CABG ranges from 24% to 57% in the first 6 months (Savageau et al. 1982; Shaw et al. 1987); one week post-operation the concentration of IL-6 in CSF is significantly increased (Kálmán et al. 2006) and does not normalize for more than six months after surgery. Overproduction of IL-6 after CABG might play a crucial role in triggering the development of cognitive dysfunction, even while still on the operating table, and elevated levels immediately after surgery may reflect increased risk of cognitive changes and the development of focal neurological deficits, e.g., stroke. A prospective observational study on 2632 elderly people by Yaffe et al. (2004), investigated the association of metabolic syndrome (a clustering of several commonly occurring disorders including hypertension, abdominal obesity, hypertriglyceridemia, low high-density lipoprotein (HDL) levels, and hyperglycaemia) and high IL-6 with a change in cognition after three and five years. Subjects with the metabolic syndrome were more likely to have cognitive impairment, and individuals with both metabolic syndrome and a high IL-6 blood concentration had a greater 4-year cognitive decline compared to those with only metabolic syndrome (Yaffe et al. 2004). A cross-sectional analysis performed in more than 5000 older individuals (the PROSPER study) with a history of cardiovascular disease showed that higher IL-6 concentration in blood is associated with an increased rate of cognitive disfunction (Mooijaart et al. 2013). Alexander et al. (2014) demonstrated that high concentration of IL-6 in blood predicts delirium or coma and thus, a higher mortality risk in severely ill patients. Recently, IL-6 overexpression in blood has been related to cognitive decline after adjusting for vascular risk factors in older individuals (Economos et al. 2013).
IL-6 and cognitive impairment in liver disease
During liver disease a wide range of pro-inflammatory markers are released into the blood stream. The concentration of IL-6 in blood serum has been positively correlated to cognitive impairment both in patients with liver cirrhosis (Montoliu et al. 2009; Jain et al. 2012; Luo et al. 2012) (Table 2) and in an animal model of hepatic encephalopathy (Cauli et al. 2007). Cirrhotic patients with IL-6 levels lower than 11 pg/ml do not show cognitive dysfunction, termed minimal hepatic encephalopathy (MHE) whereas patients with IL-6 levels above 11 pg/ml show MHE (Montoliu et al. 2009). IL-6 has good sensitivity (82 %) and specificity of 71 % suggesting that it’s a good biomarker of cognitive dysfunction in these patients. Logistic regression analysis using the presence of MHE as dependent variable and IL-6 showed that has predictive value to detect MHE (Montoliu et al. 2011). Interestingly in cirrhotic patients, the presence of moderate levels of both hyperammonemia and inflammation are required for induction of mild cognitive impairment whereas in inflammation or hyperammonemia alone does not, thus suggesting a synergism between two pathophysiological factors in the mediation of cognitive impairment. It cannot be ruled out that other types of cognitive function could be altered by hyperammonemia or inflammation alone, but impairment of the type of cognitive function assessed by the psychometric hepatic encephalopathy score (PHES) battery (the gold-standard in the detection of MHE) seem to require some grades of both hyperammonemia and inflammation. The exact mechanism by which IL-6 can induce cognitive impairment in liver cirrhosis is unknown, but several studies have provided plausible explanations. IL-6 has been reported by Weaver et al. (2002) to correlate with a cognitive decline in older people and Krabbe et al. (2005) showed a negative association between IL-6 serum levels and memory deficits during low-dose endotoxemia. Furthermore, IL-6 can alter the signal transduction related to some neurotransmitter receptors, and in particular, chronic exposure to IL-6 alters metabotropic glutamate receptor-activated calcium signalling in cerebellar neurons (Nelson et al. 2004).
IL-6 in Behçet’s disease
Behçet’s disease (BD) is a multisystem inflammatory/autoimmune disorder characterized by recurrent oral and genital ulcers, skin lesions, and relapsing uveitis (Inaba 1984; Sakane et al. 1999; Hirohata and Kikuchi 2003). A large percentage of patients with BD show a wide range of neurological problems (neuro-Behçet’s syndrome), indicating central nervous system involvement (Akman-Demir et al. 1999). The most predominant neuro-Behçet’s syndrome involves the parenchyma, brainstem, and/or spinal cord, with neuropsychological testing showing memory disturbances and cognitive impairment in most patients, and impairment to learning and recall processes in both verbal and visual modalities (Oktem-Tanör et al. 1999; Akman-Demir et al. 1999; Monastero et al. 2004; Cavaco et al. 2009). This cognitive impairment tends to worsen with time, independently of acute attacks (Oktem-Tanör et al. 1999), and is associated with poor outcome (Akman-Demir et al. 1999). Additionally, neuro-Behçet’s patients show frontal-executive dysfunction (Dutra et al. 2012; Gündüz et al. 2012) which may help explain the cognitive and behavioural (personality change) deficits often associated with this disease because these deficits are secondary to frontal and temporal cortex dysfunction caused by subcortical damage in the brainstem or basal ganglia (Arai et al. 1994; Monastero et al. 2004). Cognitive impairment has also been demonstrated in a subgroup of BD patients regardless of their overt neurological manifestations (Monastero et al. 2004; Dutra et al. 2012). Similarities between neuro-BD and BD neuropsychological alterations suggest that there is a spectrum of cognitive impairment with unknown pathophysiological origin, although accumulating evidence suggests that IL-6 is the most important cytokine in its pathogenesis (Hirohata and Kikuchi 2012). Although IL-6 serum levels first appear normal in these patients (Sayinalp et al. 1996; Yamakawa et al. 1996; Hirohata et al. 1997), during acute attacks it is released by monocytes, blood mononuclear cells, or LPS stimulation, resulting in acute inflammatory reactions (Mege et al. 1993; Yamakawa et al. 1996). In periphery, serum IL-6 appears normal in BD patients (Sayinalp et al. 1996; Yamakawa et al. 1996; Hirohata et al. 1997), while its release by monocytes, blood mononuclear cells or LPS-stimulated release is increased (Mege et al. 1993; Yamakawa et al. 1996). Interestingly significant differences in the variable number of tandem repeat polymorphisms in the 3’ flanking region of the Il-6 gene and allele frequencies were found between Korean patients with BD and controls, and susceptibility to BD significantly increased in subjects carrying some of the allele and haplotype variants (Chang et al. 2005). The IL-6 CSF concentration is also increased in patients with progressive neuro-BD, which is characterised by dementia in 82% of cases (Hirohata et al. 1997; Akman-Demir et al. 2008; Borhani Haghighi et al. 2009). Interestingly, CSF IL-6 and serum IL-6 concentrations do not significantly correlate, suggesting that high CSF IL-6 levels might result from increased intrathecal IL-6 production (Hirohata et al. 1997). The most common neuropathological feature of chronic neuro-BD is lymphocyte and proliferating microglial cell perivascular infiltration, which is thought to contribute to dementia by cytokine (including IL-6) overproduction. Indeed, several recent reports have shown that tocilizumab, an antibody against soluble and membrane-bound IL-6 receptors, has beneficial effects in neuro-BD patients (Shapiro et al. 2012; Urbaniak et al. 2012). Importantly, some patients without any overt neurological manifestation may have increased CSF IL-6 levels, sometimes associated with subtle, subclinical cognitive impairment (Hirohata et al. 1997).
IL-6 in Systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is an autoimmune disease characterised by deregulation of the immune system including T- and B-lymphocyte hyperactivity, production of autoantibodies, and the formation of immune complex deposits in tissues causing organ damage. A wide range of nervous system diseases are also associated with SLE (Hanly et al. 2007) and importantly, CNS involvement is associated with increased morbidity and mortality. Cognitive impairment is a common neuropsychiatric (NP) event in patients with SLE; there is no specific pattern of cognitive dysfunction in SLE, but characteristics include overall cognitive slowing, decreased attention, impaired working memory, and executive dysfunction (e.g., difficulty with multitasking, organization, and planning). Hence the American College of Rheumatology (ACR) has proposed a battery of neuropsychological tests to assess the cognitive function of SLE patients (ACR 1999; Lim et al. 2013). The majority of SLE patients have subtle, frequently subclinical, cognitive deficits without any overt neuropsychiatric symptoms (stroke, seizures, organic brain syndrome, non-organic psychosis, chorea, focal lesions, or psychosis/major depression), although profound symptoms sometimes present in individual cases (Hay et al. 1994; Hanly et al. 1997; Waterloo et al. 2002; Kozora et al. 2012; Hanly et al. 2012). Mild cognitive impairment, characterized by visual/complex attention, visuomotor speed, and verbal memory impairment, and the absence of overt neuropsychiatric symptoms, has recently been described in 20-30% of SLE patients (Kozora et al. 2004; Kozora et al. 2012).
Serum IL-6 concentrations greatly increase in patients with SLE during acute phases of the disease (Linker-Israeli et al. 1991; Spronk et al. 1992; Davas et al. 1999; Chung et al. 2007), however measurements of peripheral blood IL-6 levels in stable disease conditions yielded mixed results (Hirohata and Miyamoto 1990; Kozora et al. 2004; Kozora et al. 2012). Trysberg et al. (2000) reported that serum IL-6 did not differ between SLE patients with and without neuropsychiatric symptoms; another study found no relationship between increased peripheral blood IL-6 levels and cognitive impairment in SLE patients without overt neuropsychiatric symptoms (Kozora et al. 2012). The concentration of IL-6 in the CSF is increased in SLE patients with these symptoms (Hirohata and Miyamoto 1990; Trysberg et al. 2000; Dellalibera-Joviliano et al. 2003), but thus far cognitive impairment has not been correlated with CSF IL-6 concentrations in SLE patients. CSF IL-6 levels do not correlate with serum IL-6 content nor CSF serum albumin quotients, suggesting that IL-6 overproduction occurs in these patient’s brains (Hirohata and Miyamoto 1990; Trysberg et al. 2000). Moreover, remission of overt neuropsychiatric manifestations after successful treatment is accompanied by a reduction of CSF IL-6 activity (Hirohata and Miyamoto 1990).
Discussion
We will discuss the most important conclusions separately. First addressing how IL-6 leads to cognitive impairment, and then we discuss our main conclusions related to each pathology reviewed in this manuscript.
Molecular mechanisms by which IL-6 overproduction leads to cognitive impairment
The effects of IL-6 in the central nervous system (CNS) are quite complex and diverse, and the mechanisms by which IL-6 influences neuronal functions and thus cognitive function are primarily unknown (Jüttler et al. 2002). IL-6 overexpression may induce cognitive dysfunction by impairing neurotransmission in brain structures which modulate cognitive functions e.g. the hippocampus and prefrontal cortex. In support of this, the concentration of 5-hydroxyindole acetic acid (5-HIAA), a major serotonin (5-hydroxytryptamine; 5-HT) metabolite, increases in these brain areas in IL-6-administered mice (Zalcman et al. 1994; Wang and Dunn 1998). Furthermore, intracerebroventricular administration of human IL-6 increases extracellular hypothalamic 5-HT concentrations (Wu et al. 1999), while intraperitoneal recombinant IL-6 injection elevates microdialysate 5-HT concentrations in the striatum (Zhang et al. 2001), enhances 5-HT-like signals from the striatum following electrical stimulation of the dorsal raphe nucleus. Additionally, these results indicate that the increases in brain concentrations of 5-HIAA observed in earlier studies indeed reflect increased synaptic release of 5-HT (Zhang et al. 2001). One can speculate that increased 5-HT release induced by IL-6 is secondary to activation of the hypothalamic-pituitary-adrenal axis and release of corticosterone (Naitoh et al. 1988), however other studies may exclude this possibility because administration of corticosterone failed to alter brain tryptophan and 5-HIAA:5-HT ratios in mice (Dunn 1992), and IL-6 only increases plasma cortiscosterone concentrations when administered at doses similar to levels present during acute infections (Dunn 1992; Zalcman et al. 1994; Wang and Dunn 1998). IL-6 increases dopamine (DA) turnover in the prefrontal cortex but does not affect DA or noradrenaline concentrations in other brain regions (Zalcman et al. 1994; Wang and Dunn 1998). Several in vitro studies have demonstrated that IL-6 impairs some excitatory synaptic activity. Indeed, IL-6 is required to regulate the expression of functional hippocampal and cortical adenosine A1 receptors, i.e. exposure to IL-6 potentiates adenosine A1 receptors to inhibit excitatory synaptic transmissions in hippocampal slices (Biber et al. 2008). Additionally IL-6 depresses the spread of excitation and evokes glutamate release in the cerebral cortex by downregulating neuronal activity, releasing excitatory neurotransmitters, activating MAPK/ERK activity, and stimulating neuron-expressed IL-6-receptor signal transduction pathways (D'Arcangelo et al. 2000). IL-6 can also impair synaptic transmission by decreasing post-tetanic potentiation and long-term potentiation (Tancredi et al. 2000) and selectively and reversibly reduces the amplitude of inhibitory postsynaptic currents in cortical rat brain slices by shifting the balance from synaptic inhibition to excitation. These decreases in inhibitory synaptic activity appear to result from a reduction of functional GABA-A receptors, mediated by their accelerated removal and/or a decrease in their insertion rate into the plasma membrane (Garcia-Oscos et al. 2012). IL-6 also selectively disrupts cholinergic transmission by inducing functional pathophysiology in hippocampal cholinergic target neurons (Steffensen et al. 1994) and by altering metabotropic glutamate receptor-activated calcium signaling (Nelson et al. 2004).
Taken together this experimental evidence suggests that IL-6-induced alterations in monoamine (mainly 5-HT) concentrations and the release of synaptic activity in the hippocampus and frontal cortex (through glutamatergic and GABA-ergic neurotransmission), may contribute to the cytokine's effects on cognitive function (Fig. 2).

Schematic representation of IL-6 actions related to cognitive impairment. An increase synthesis of IL-6 in the brain (from microglia cells, astrocytes, neurons oand endothelial cells) or an increased uptake from blood leads to cognitive impairment by altering serotonin neurotransmission, synaptic activity by altering the function of several neurotransmitters and their receptors or impairing neurogenesis. Glu: glutamate; GABA; γ-aminobutirric acid; Ach: acetylcholine; ADO: adenosine
Another mechanism by which IL-6 can lead to cognitive dysfunction is the impairment of adult hippocampal neurogenesis. Several inflammation factors, especially IL6, are detrimental for neurogenesis (Vallières et al. 2002; Monje et al. 2003). In addition, an inverse relationship between blood IL-6 concentration and hippocampal grey matter volume have been found (Marsland et al. 2008). A recent paper even showed blockade of IL6 in a mouse model with loss of neurogenesis could largely restore hippocampal neurogenesis (Ji et al. 2013) suggesting for the first time that peripheral IL-6 can impair cognitive function via structural neural pathways.
Is IL-6 overexpression associated with cognitive impairment?
Here we discuss our conclusions separately because they vary depending on the disease considered.
Alzheimer’s disease and vascular dementia
Increased IL-6 production in the brain has been repeatedly reported in AD, with the main sources being reactive astrocytosis and activation of microglia. This increase appears to be related to early-stage plaque formation (Huell et al. 1995; Jellinger 2009) and is most relevant in those patients with late onset AD. However further studies should address if changes in IL-6 concentration are significantly correlated with cognitive performance in AD patients as it is in patients with mild cognitive impairment (Sala et al. 2003; Rafnsson et al. 2007; Zhao et al. 2012). Measurements of plasma or serum IL-6 concentration in AD patients have yielded mixed results and no fixed conclusions can be drawn because several factors such as AD subtype (early versus late onset), age, sex, and genetic IL-6 polymorphisms may account for these discrepancies. However, it appears that IL-6 might be involved in the pathogenesis of AD, but that the IL-6 CSF concentration is unlikely to affect AD pathology in the early stages of the disorder. Systemically-produced IL-6 may permeate across the BBB and, after reaching the brain, might contribute to the neuropathology in patients with mild cognitive impairment that progresses to AD. In contrast to AD, there is a consensus in the literature on the increase in brain and serum IL-6 concentration in vascular dementia (VD). Interestingly plasma IL-6 correlated with the functional status of patients (Barthel score) and to some extent also to their cognitive performance (Zuliani et al. 2007a; Zuliani et al. 2007b).
Liver disease and cardiovascular disease
Several pathologies and syndromes manifest with chronic inflammation and overproduction of IL-6 in the blood. Peripheral IL-6 concentration has been consistently found to be increased in patients with alcoholic liver cirrhosis and the increase significantly correlates with the impairment of cognitive functions (Montoliu et al. 2009; Jain et al. 2012; Luo et al. 2012). Similarly, cognitive impairment displayed by patients after hearth stroke is also significantly correlated with a poorer score in the mini-mental examination test (Rothenburg et al. 2010). Therefore the data suggest that peripheral inflammation, characterised by an increase in blood IL-6 concentration, is frequently accompanied by parallel cognitive impairment in patients with peripheral disease although the mechanisms are poorly understood. Older patients with metabolic syndrome (characterized by hypertension, obesity, hyperglycaemia, and high cholesterol/triglycerides) and a high blood concentration of IL-6 have greater cognitive decline compared to those with normal levels of IL-6 (Yaffe et al. 2004).
Behçet’s disease and systemic lupus erythematosus
In patients with inflammatory diseases such as BD IL-6 this has also been linked to cognitive impairment, at least in a subgroup of patients with neuro-BD which manifests independently of acute inflammatory attacks, which is associated with poor outcome ranging in severity from subtle cognitive impairment to clinical forms of dementia (Hirohata et al. 1997; Akman-Demir et al. 1999; Borhani Haghighi et al. 2009); (Oktem-Tanör et al. 1999; Akman-Demir et al. 1999; Hirohata and Kikuchi 2012); (Hirohata et al. 1997; Monastero et al. 2004; Akman-Demir et al. 2008; Borhani Haghighi et al. 2009; Dutra et al. 2012). Increased in IL-6 concentrations in CSF have also been shown in patients with neuro-BD and dementia (Hirohata et al. 1997) and therefore it has been proposed that it CSF IL-6 could be used as marker of disease activity in chronic progressive patients with neuro-BD (Akman-Demir et al. 2008). The reason why only some BD patients display increased IL-6 and cognitive dysfunction warrants further investigation, taking into account age, IL-6 and IL-6 R polymorphisms (Chang et al. 2005; Amirzargar et al. 2010). However the idea that an increase in CSF IL-6 is associated with subtle, sub-clinical, cognitive impairment in BD patients without any overt neurological manifestation cannot yet be discarded. A high percentage of patients with SLE show cognitive dysfunction even in the absence of overt neurological manifestations (Waterloo et al. 2002; Kozora et al. 2012; Hanly et al. 2012). Increased IL-6 CSF concentrations have also been reported in SLE patients (Hirohata and Miyamoto 1990; Trysberg et al. 2000), although these findings still require further exploration as one study found no such correlation with cognitive impairment (Kozora et al. 2012). However this discrepancy could be explained by an age and gender bias as SLE is more common in females and the SLE patients enrolled in the study in question were younger than average. Finally, serum IL-6 is not always increased in SLE patients (Hirohata and Miyamoto 1990; Kozora et al. 2004; Kozora et al. 2012) suggesting that disease phases (e.g. acute attacks versus chronic stable conditions) should be also evaluated when assessing the role of peripheral IL-6 in cognitive impairment. An evaluation of the relationship between cognitive dysfunction and changes in inflammatory markers over a 9-year period found that a 2-fold increase in IL-6 is a good predictor of cognitive impairment (Jenny et al. 2012).
Other diseases
The concentration of IL-6 in blood and in the brain is also increased in other neuropsychiatric diseases such as Huntington's disease (Silvestroni et al. 2009; Sánchez-López et al. 2012), major depression (Rosenblat et al. 2014), schizophrenia (Stojanovic et al. 2014) and autism (Wei et al. 2013) but data about the releationship between IL-6 and cognitive impairment in these diseases have not been reported.
Concluding remarks and new therapeutic possibilities
Future studies should always aim to determine both CSF and blood IL-6 concentrations because the presence or absence of IL-6 overproduction in these different fluids implies the involvement of different processes. IL-6 overproduction must occur in the brain if brain dysfunction is induced, but increased peripheral IL-6 overproduction also directly or indirectly results in a rise in IL-6 concentration in the brain. Although peripheral IL-6 is useful for indicating a proinflammatory disease state or an acute phase response, it is important to consider that the presence of specific circulating markers may not precisely reflect the situation within the CNS. Finally, evaluation of cognitive function in pathologies which are accompanied by a strong increase of IL-6 in the blood (e.g. Castleman’s Disease, POEMS syndrome, some cancers etc.) is advisable since it will shed new light on the role of peripheral IL-6 in cognitive dysfunction. Thus, although the importance of IL-6 in cognitive processes has been suggested in several pathologies, as this review discusses, the relevance of subtle rises in serum IL-6 (secondary to infections and/or inflammation) to cognitive outcomes remains unclear and warrants further detailed study.
Identifying blood markers that distinguish cognitive impairment, or the severity of cognitive dysfunction within the same diagnostic group, would be useful for defining disease etiology, devising new therapies, or finding ways to prevent it. Identifying such peripheral biomarkers would be very helpful in pinpointing specific causal mechanisms and may result in tailored treatments or prevention strategies. Future studies will need to address whether lowering inflammation responses prevents cognitive impairment in systemic diseases.
References
ACR (1999) The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheum 42:599–608
Akaneya Y, Takahashi M, Hatanaka H (1995) Interleukin-1 beta enhances survival and interleukin-6 protects against MPP+ neurotoxicity in cultures of fetal rat dopaminergic neurons. Exp Neurol 136:44–52
Akiyama H, Barger S, Barnum S et al (2000) Inflammation and Alzheimer's disease. Neurobiology of Aging 21:383–421
Akman-Demir G, Serdaroglu P, Tasçi B (1999) Clinical patterns of neurological involvement in Behçet's disease: evaluation of 200 patients. The Neuro-Behçet Study Group Brain 122(Pt 11):2171–2182
Akman-Demir G, Tüzün E, Içöz S et al (2008) Interleukin-6 in neuro-Behçet's disease: association with disease subsets and long-term outcome. Cytokine 44:373–376
Alexander SA, Ren D, Gunn SR et al (2014) Interleukin 6 and apolipoprotein E as predictors of acute brain dysfunction and survival in critical care patients. Am J Crit Care 23:49–57
Amirzargar A, Shahram F, Nikoopour E et al (2010) Proinflammatory cytokine gene polymorphisms in Behçet's disease. Eur Cytokine Netw 21:292–296
Amos A (2002) Remediating deficits of switching attention in patients with acquired brain injury. Brain Inj 16:407–413
Andriambeloson E, Baillet C, Vitte P-A et al (2006) Interleukin-6 attenuates the development of experimental diabetes-related neuropathy. Neuropathology 26:32–42
Angelis P, Scharf S, Mander A et al (1998) Serum interleukin-6 and interleukin-6 soluble receptor in Alzheimer's disease. Neurosci Lett 244:106–108
Arai T, Mizukami K, Sasaki M et al (1994) Clinicopathological study on a case of neuro-Behçet's disease: in special reference to MRI, SPECT and neuropathological findings. Jpn J Psychiatry Neurol 48:77–84
Barres BA, Schmid R, Sendnter M, Raff MC (1993) Multiple extracellular signals are required for long-term oligodendrocyte survival. Development 118:283–295
Baskys A, Cheng J-X (2012) Pharmacological prevention and treatment of vascular dementia: approaches and perspectives. Experimental Gerontology 47:887–891
Bauer J, Ganter U, Strauss S et al (1992) The participation of interleukin-6 in the pathogenesis of Alzheimer's disease. Res Immunol 143:650–657
Berkenbosch F, Biewenga J, Brouns M et al (1992) Cytokines and inflammatory proteins in Alzheimer's disease. Res Immunol 143:657–663
Bermejo P, Martín-Aragón S, Benedí J et al (2008) Differences of peripheral inflammatory markers between mild cognitive impairment and Alzheimer's disease. Immunology Letters 117:198–202
Biber K, Pinto-Duarte A, Wittendorp MC et al (2008) Interleukin-6 upregulates neuronal adenosine A1 receptors: implications for neuromodulation and neuroprotection. Neuropsychopharmacology 33:2237–2250
Blum-Degen D, Müller T, Kuhn W et al (1995) Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer“s and de novo Parkinson”s disease patients. Neurosci Lett 202:17–20
Borhani Haghighi A, Ittehadi H, Nikseresht AR et al (2009) CSF levels of cytokines in neuro-Behçet's disease. Clin Neurol Neurosurg 111:507–510
Borowicz LM, Goldsborough MA, Selnes OA, McKhann GM (1996) Neuropsychologic change after cardiac surgery: a critical review. J Cardiothorac Vasc Anesth 10:105–112
Boulanger MJ, Chow D-C, Brevnova EE, Garcia KC (2003) Hexameric Structure and Assembly of the Interleukin-6/IL-6 {alpha}-Receptor/gp130 Complex. Science Signaling 300:2101–2104
Cauli O, Rodrigo R, Piedrafita B et al (2007) Inflammation and hepatic encephalopathy: ibuprofen restores learning ability in rats with portacaval shunts. Hepatology 46:514–519
Cavaco S, da Silva AM, Pinto P et al (2009) Cognitive functioning in Behçet's disease. Ann N Y Acad Sci 1173:217–226
Cercy SP, Bylsma FW (1997) Lewy bodies and progressive dementia: a critical review and meta-analysis. J Int Neuropsychol Soc 3:179–194
Cesari M, Penninx BWJH, Pahor M et al (2004) Inflammatory markers and physical performance in older persons: the InCHIANTI study. J Gerontol A Biol Sci Med Sci 59:242–248
Chang HK, Jang WC, Park SB et al (2005) Association between interleukin 6 gene polymorphisms and Behcet's disease in Korean people. Ann Rheum Dis 64:339–340
Chow D-C, He X-L, Snow AL et al (2001) Structure of an Extracellular gp130 Cytokine Receptor Signaling Complex. Science Signaling 291:2150–2155
Chung CP, Oeser A, Solus J et al (2007) Inflammatory mechanisms affecting the lipid profile in patients with systemic lupus erythematosus. J Rheumatol 34:1849–1854
Cojocaru IM, Cojocaru M, Miu G, Sapira V (2011) Study of interleukin-6 production in Alzheimer's disease. Rom J Intern Med 49:55–58
D'Arcangelo G, Tancredi V, Onofri F et al (2000) Interleukin-6 inhibits neurotransmitter release and the spread of excitation in the rat cerebral cortex. Eur J Neurosci 12:1241–1252
Davas EM, Tsirogianni A, Kappou I et al (1999) Serum IL-6, TNFalpha, p55 srTNFalpha, p75srTNFalpha, srIL-2alpha levels and disease activity in systemic lupus erythematosus. Clin Rheumatol 18:17–22
De Simoni MG, De Luigi A, Gemma L et al (1993) Modulation of systemic interleukin-6 induction by central interleukin-1. Am J Physiol 265:R739–42
Dellalibera-Joviliano R, Reis Dos ML, Cunha F, De Q, Donadi EA (2003) Kinins and cytokines in plasma and cerebrospinal fluid of patients with neuropsychiatric lupus. J Rheumatol 30:485–492
Dendorfer U, Oettgen P, Libermann TA (1994) Multiple regulatory elements in the interleukin-6 gene mediate induction by prostaglandins, cyclic AMP, and lipopolysaccharide. Mol Cell Biol 14:4443–4454
Di Santo E, Alonzi T, Fattori E et al (1996) Overexpression of interleukin-6 in the central nervous system of transgenic mice increases central but not systemic proinflammatory cytokine production. Brain Res 740:239–244
Dunn AJ (1992) The role of interleukin-1 and tumor necrosis factor alpha in the neurochemical and neuroendocrine responses to endotoxin. Brain Res Bull 29:807–812
Dutra LA, Gonçalves CR, Braga-Neto P et al (2012) Atypical manifestations in Brazilian patients with neuro-Behçet's disease. J Neurol 259:1159–1165
Economos A, Wright CB, Moon YP et al (2013) Interleukin 6 Plasma Concentration Associates with Cognitive Decline: The Northern Manhattan Study. Neuroepidemiology 40:253–259
Egashira K (2003) Molecular mechanisms mediating inflammation in vascular disease: special reference to monocyte chemoattractant protein-1. Hypertension 41:834–841
Engelhart MJ, Geerlings MI, Meijer J et al (2004) Inflammatory proteins in plasma and the risk of dementia: the rotterdam study. Arch Neurol 61:668–672
Ershler WB, Keller ET (2000) Age-associated increased interleukin-6 gene expression, late-life diseases, and frailty. Annu Rev Med 51:245–270
Fried LP, Guralnik JM (1997) Disability in older adults: evidence regarding significance, etiology, and risk. J Am Geriatr Soc 45:92–100
Gadient RA, Otten U (1994a) Identification of interleukin-6 (IL-6)-expressing neurons in the cerebellum and hippocampus of normal adult rats. Neurosci Lett 182:243–246
Gadient RA, Otten U (1993) Differential expression of interleukin-6 (IL-6) and interleukin-6 receptor (IL-6R) mRNAs in rat hypothalamus. Neurosci Lett 153:13–16
Gadient RA, Otten U (1994b) Expression of interleukin-6 (IL-6) and interleukin-6 receptor (IL-6R) mRNAs in rat brain during postnatal development. Brain Res 637:10–14
Gadient RA, Otten UH (1997) Interleukin-6 (IL-6)–a molecule with both beneficial and destructive potentials. Prog Neurobiol 52:379–390
Garcia-Oscos F, Salgado H, Hall S et al (2012) The stress-induced cytokine interleukin-6 decreases the inhibition/excitation ratio in the rat temporal cortex via trans-signaling. Biological Psychiatry 71:574–582
Gauldie J, Sauder DN, McAdam KP, Dinarello CA (1987) Purified interleukin-1 (IL-1) from human monocytes stimulates acute-phase protein synthesis by rodent hepatocytes in vitro. Immunology 60:203–207
Girotti M, Donegan JJ, Morilak DA (2012) Influence of hypothalamic IL-6/gp130 receptor signaling on the HPA axis response to chronic stress. Psychoneuroendocrinology 38:1158–1169
Goodman MN (1994) Interleukin-6 induces skeletal muscle protein breakdown in rats. Proc Soc Exp Biol Med 205:182–185
Gómez-Tortosa E, Gonzalo I, Fanjul S et al (2003) Cerebrospinal fluid markers in dementia with lewy bodies compared with Alzheimer disease. Arch Neurol 60:1218–1222
Gruol DL, Nelson TE (1997) Physiological and pathological roles of interleukin-6 in the central nervous system. Mol Neurobiol 15:307–339
Guzmán C, Hallal-Calleros C (2010) Interleukin-6: a cytokine with a pleiotropic role in the neuroimmunoendocrine network. The Open Neuroendocrinology Journal 3:152–160
Gündüz T, Emir Ö, Kürtüncü M et al (2012) Cognitive impairment in neuro-Behcet's disease and multiple sclerosis: a comparative study. Int J Neurosci 122:650–656
Hama T, Kushima Y, Miyamoto M et al (1991) Interleukin-6 improves the survival of mesencephalic catecholaminergic and septal cholinergic neurons from postnatal, two-week-old rats in cultures. Neuroscience 40:445–452
Hammeke TA, Hastings JE (1988) Neuropsychologic alterations after cardiac operation. J Thorac Cardiovasc Surg 96:326–331
Hampel H, Haslinger A, Scheloske M et al (2005) Pattern of interleukin-6 receptor complex immunoreactivity between cortical regions of rapid autopsy normal and Alzheimer's disease brain. Eur Arch Psychiatry Clin Neurosci 255:269–278
Hanly JG, Cassell K, Fisk JD (1997) Cognitive function in systemic lupus erythematosus: results of a 5-year prospective study. Arthritis Rheum 40:1542–1543
Hanly JG, Su L, Omisade A et al (2012) Screening for cognitive impairment in systemic lupus erythematosus. J Rheumatol 39:1371–1377
Hanly JG, Urowitz MB, Sanchez-Guerrero J et al (2007) Neuropsychiatric events at the time of diagnosis of systemic lupus erythematosus: an international inception cohort study. Arthritis Rheum 56:265–273
Hay EM, Huddy A, Black D et al (1994) A prospective study of psychiatric disorder and cognitive function in systemic lupus erythematosus. Ann Rheum Dis 53:298–303
Heinrich PC, Behrmann I, Müller-Newen G et al (1998) Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J 334(Pt 2):297–314
Henriksen L, Hjelms E, Lindeburgh T (1983) Brain hyperperfusion during cardiac operations. Cerebral blood flow measured in man by intra-arterial injection of xenon 133: evidence suggestive of intraoperative microembolism. J Thorac Cardiovasc Surg 86:202–208
Hirohata S (1996) Coping with pathological changes in nerves and blood vessels in collagen disease–CNS lupus. Nippon Naika Gakkai Zasshi 85:1816–1821
Hirohata S, Isshi K, Oguchi H et al (1997) Cerebrospinal fluid interleukin-6 in progressive Neuro-Behçet's syndrome. Clin Immunol Immunopathol 82:12–17
Hirohata S, Kikuchi H (2003) Behçet's disease. Arthritis Res Ther 5:139–146
Hirohata S, Kikuchi H (2012) Changes in biomarkers focused on differences in disease course or treatment in patients with neuro-Behçet's disease. Intern Med 51:3359–3365
Hirohata S, Miyamoto T (1990) Elevated levels of interleukin-6 in cerebrospinal fluid from patients with systemic lupus erythematosus and central nervous system involvement. Arthritis Rheum 33:644–649
Huell M, Strauss S, Volk B et al (1995) Interleukin-6 is present in early stages of plaque formation and is restricted to the brains of Alzheimer's disease patients. Acta Neuropathol 89:544–551
Hull M, Berger M, Volk B, Bauer J (1996a) Occurrence of interleukin-6 in cortical plaques of Alzheimer's disease patients may precede transformation of diffuse into neuritic plaques. Ann N Y Acad Sci 777:205–212
Hull M, Strauss S, Berger M et al (1996b) Inflammatory mechanisms in Alzheimer's disease. Eur Arch Psychiatry Clin Neurosci 246:124–128
Imamura K, Hishikawa N, Ono K et al (2004) Cytokine production of activated microglia and decrease in neurotrophic factors of neurons in the hippocampus of Lewy body disease brains. Acta Neuropathol 109:141–150
Inaba G (1984) Revised criteria for the diagnosis of Behçet's disease. Ryumachi 24:135–141
Jain L, Sharma BC, Sharma P et al (2012) Serum endotoxin and inflammatory mediators in patients with cirrhosis and hepatic encephalopathy. Dig Liver Dis 44:1027–1031
Jellinger KA (2009) Intrathecal levels of IL-6 in Alzheimer’s disease. J Neurol 257:142–142
Jenny NS, French B, Arnold AM et al (2012) Long-term assessment of inflammation and healthy aging in late life: the Cardiovascular Health Study All Stars. J Gerontol A Biol Sci Med Sci 67:970–976
Ji R, Tian S, Lu HJ et al (2013) TAM receptors affect adult brain neurogenesis by negative regulation of microglial cell activation. J Immunol 191:6165–6177
Jia JP, Meng R, Sun YX et al (2005) Cerebrospinal fluid tau, Abeta1-42 and inflammatory cytokines in patients with Alzheimer's disease and vascular dementia. Neurosci Lett 383:12–16
Jüttler E, Tarabin V, Schwaninger M (2002) Interleukin-6 (IL-6): a possible neuromodulator induced by neuronal activity. Neuroscientist 8:268–275
Kalman J, Juhasz A, Laird G et al (2009) Serum interleukin-6 levels correlate with the severity of dementia in Down syndrome and in Alzheimer's disease. Acta Neurol Scand 96:236–240
Kalman J, Juhasz A, Laird G et al (1997) Serum interleukin-6 levels correlate with the severity of dementia in Down syndrome and in Alzheimer's disease. Acta Neurol Scand 96:236–240
Kálmán J, Juhász A, Bogáts G et al (2006) Elevated levels of inflammatory biomarkers in the cerebrospinal fluid after coronary artery bypass surgery are predictors of cognitive decline. Neurochemistry International 48:177–180
Kent SS, Kelley KWK, Dantzer RR (1992) Effects of lipopolysaccharide on food-motivated behavior in the rat are not blocked by an interleukin-1 receptor antagonist. Neurosci Lett 145:83–86
Klein MA, Möller JC, Jones LL et al (1997) Impaired neuroglial activation in interleukin-6 deficient mice. Glia 19:227–233
Kopf M, Baumann H, Freer G et al (1994) Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature 368:339–342
Kossmann T, Hans V, Imhof HG et al (1996) Interleukin-6 released in human cerebrospinal fluid following traumatic brain injury may trigger nerve growth factor production in astrocytes. Brain Res 713:143–152
Kozora E, Ellison MC, West S (2004) Reliability and validity of the proposed American College of Rheumatology neuropsychological battery for systemic lupus erythematosus. Arthritis Rheum 51:810–818
Kozora E, Filley CM, Zhang L et al (2012) Immune function and brain abnormalities in patients with systemic lupus erythematosus without overt neuropsychiatric manifestations. Lupus 21:402–411
Krabbe KS, Pedersen M, Bruunsgaard H (2004) Inflammatory mediators in the elderly. Experimental Gerontology 39:687–699
Krabbe KS, Reichenberg A, Yirmiya R et al (2005) Low-dose endotoxemia and human neuropsychological functions. BRAIN, BEHAVIOUR, AND IMMUNITY 19:453–460
Kushima Y, Hama T, Hatanaka H (1992) Interleukin-6 as a neurotrophic factor for promoting the survival of cultured catecholaminergic neurons in a chemically defined medium from fetal and postnatal rat midbrains. Neurosci Res 13:267–280
Kushima Y, Hatanaka H (1992) Interleukin-6 and leukemia inhibitory factor promote the survival of acetylcholinesterase-positive neurons in culture from embryonic rat spinal cord. Neurosci Lett 143:110–114
Lanzrein AS, Johnston CM, Perry VH et al (1998) Longitudinal study of inflammatory factors in serum, cerebrospinal fluid, and brain tissue in Alzheimer disease: interleukin-1beta, interleukin-6, interleukin-1 receptor antagonist, tumor necrosis factor-alpha, the soluble tumor necrosis factor receptors I and II, and alpha1-antichymotrypsin. Alzheimer Dis Assoc Disord 12:215–227
Licastro F, Grimaldi LME, Bonafè M et al (2003) Interleukin-6 gene alleles affect the risk of Alzheimer's disease and levels of the cytokine in blood and brain. NBA 24:921–926
Licastro F, Pedrini S, Caputo L et al (2000) Increased plasma levels of interleukin-1, interleukin-6 and alpha-1-antichymotrypsin in patients with Alzheimer's disease: peripheral inflammation or signals from the brain? Journal of Neuroimmunology 103:97–102
Lim L, Lippe S, Silverman E (2013) Effect of autoimmune diseases on cognitive function. Handb Clin Neurol 112:1275–1283
Linker-Israeli M, Deans RJ, Wallace DJ et al (1991) Elevated levels of endogenous IL-6 in systemic lupus erythematosus. A putative role in pathogenesis J Immunol 147:117–123
Luo M, Li L, Yang E-N, Cao W-K (2012) Relationship between interleukin-6 and ammonia in patients with minimal hepatic encephalopathy due to liver cirrhosis. Hepatol Res 42:1202–1210
Marsland AL, Gianaros PJ, Abramowitch SM et al (2008) Interleukin-6 Covaries Inversely with Hippocampal Grey Matter Volume in Middle-Aged Adults. Biological Psychiatry 64:484–490
Martínez M, Fernández-Vivancos E, Frank A et al (2000) Increased cerebrospinal fluid fas (Apo-1) levels in Alzheimer's disease. Relationship with IL-6 concentrations. Brain Res 869:216–219
Mastorakos G, Ilias I (2006) Interleukin-6: a cytokine and/or a major modulator of the response to somatic stress. Ann N Y Acad Sci 1088:373–381
May LT, Santhanam U, Sehgal PB (1991) On the multimeric nature of natural human interleukin-6. J Biol Chem 266:9950–9955
März P, Heese K, Hock C et al (1997) Interleukin-6 (IL-6) and soluble forms of IL-6 receptors are not altered in cerebrospinal fluid of Alzheimer's disease patients. Neurosci Lett 239:29–32
McKeith IG, Galasko D, Kosaka K et al (1996) Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology 47:1113–1124
Mege JL, Dilsen N, Sanguedolce V et al (1993) Overproduction of monocyte derived tumor necrosis factor alpha, interleukin (IL) 6, IL-8 and increased neutrophil superoxide generation in Behçet's disease. A comparative study with familial Mediterranean fever and healthy subjects J Rheumatol 20:1544–1549
Monastero R, Camarda C, Pipia C et al (2004) Cognitive impairment in Behçet's disease patients without overt neurological involvement. Journal of the Neurological Sciences 220:99–104
Montagnese S, Biancardi A, Schiff S et al (2011) Different biochemical correlates for different neuropsychiatric abnormalities in patients with cirrhosis. Hepatology 53:558–566
Monje ML, Toda H, Palmer TD (2003) Inflammatory blockade restores adult hippocampal neurogenesis. Science 302:1760–1765
Montoliu C, Cauli O, Urios A et al (2011) 3-nitro-tyrosine as a peripheral biomarker of minimal hepatic encephalopathy in patients with liver cirrhosis. Am J Gastroenterol 106:1629–1637
Montoliu C, Piedrafita B, Serra MA et al (2009) IL-6 and IL-18 in blood may discriminate cirrhotic patients with and without minimal hepatic encephalopathy. J Clin Gastroenterol 43:272–279
Mooijaart SP, Sattar N, Trompet S et al (2013) Circulating interleukin-6 concentration and cognitive decline in old age: the PROSPER study. J Intern Med 274:77–85
Murphy PG, Grondin J, Altares M, Richardson PM (1995) Induction of interleukin-6 in axotomized sensory neurons. J Neurosci 15:5130–5138
Naitoh Y, Fukata J, Tominaga T et al (1988) Interleukin-6 stimulates the secretion of adrenocorticotropic hormone in conscious, freely-moving rats. Biochem Biophys Res Commun 155:1459–1463
Nelson TE, Netzeband JG, Gruol DL (2004) Chronic interleukin-6 exposure alters metabotropic glutamate receptor-activated calcium signalling in cerebellar Purkinje neurons. Eur J Neurosci 20:2387–2400
Nybo L, Nielsen B, Pedersen BK et al (2002) Interleukin-6 release from the human brain during prolonged exercise. J Physiol (Lond) 542:991–995
Oktem-Tanör O, Baykan-Kurt B, Gürvit IH et al (1999) Neuropsychological follow-up of 12 patients with neuro-Behçet disease. J Neurol 246:113–119
Peters M, Müller AM, Rose-John S (1998) Interleukin-6 and soluble interleukin-6 receptor: direct stimulation of gp130 and hematopoiesis. Blood 92:3495–3504
Pola R, Flex A, Gaetani E et al (2002) The −174 G/C polymorphism of the interleukin-6 gene promoter and essential hypertension in an elderly Italian population. J Hum Hypertens 16:637–640
Pugsley W, Klinger L, Paschalis C et al (1994) The impact of microemboli during cardiopulmonary bypass on neuropsychological functioning. Stroke 25:1393–1399
Qiu Z, Sweeney DD, Netzeband JG, Gruol DL (1998) Chronic interleukin-6 alters NMDA receptor-mediated membrane responses and enhances neurotoxicity in developing CNS neurons. J Neurosci 18:10445–10456
Rafnsson SB, Deary IJ, Smith FB et al (2007) Cardiovascular diseases and decline in cognitive function in an elderly community population: the Edinburgh Artery Study. Psychosom Med 69:425–434
Rose-John S (2012) IL-6 trans-signaling via the soluble IL-6 receptor: importance for the pro-inflammatory activities of IL-6. Int J Biol Sci 8:1237–1247
Rosenblat JD, Cha DS, Mansur RB, McIntyre RS (2014) Inflamed moods: A review of the interactions between inflammation and mood disorders. Prog Neuropsychopharmacol Biol Psychiatry 53C:23–34
Rothenburg LS, Herrmann N, Swardfager W et al (2010) The relationship between inflammatory markers and post stroke cognitive impairment. J Geriatr Psychiatry Neurol 23:199–205
Sakane T, Takeno M, Suzuki N, Inaba G (1999) Behçet's disease. N Engl J Med 341:1284–1291
Sala G, Galimberti G, Canevari C et al (2003) Peripheral cytokine release in Alzheimer patients: correlation with disease severity. Neurobiology of Aging 24:909–914
Savageau JA, Stanton BA, Jenkins CD, Frater RW (1982) Neuropsychological dysfunction following elective cardiac operation. II A six-month reassessment J Thorac Cardiovasc Surg 84:595–600
Sawada M, Imamura K, Nagatsu T (2006) Role of cytokines in inflammatory process in Parkinson's disease. J Neural Transm Suppl 70:373–381
Sawada M, Suzumura A, Marunouchi T (1995) Cytokine network in the central nervous system and its roles in growth and differentiation of glial and neuronal cells. Int J Dev Neurosci 13:253–264
Sayinalp N, Ozcebe OI, Ozdemir O et al (1996) Cytokines in Behçet's disease. J Rheumatol 23:321–322
Sánchez-López F, Tasset I, Agüera E et al (2012) Oxidative stress and inflammation biomarkers in the blood of patients with Huntington's disease. Neurol Res 34:721–724
Schöbitz B, de Kloet ER, Holsboer F (1994) Gene expression and function of interleukin 1, interleukin 6 and tumor necrosis factor in the brain. Prog Neurobiol 44:397–432
Schöbitz B, de Kloet ER, Sutanto W, Holsboer F (1993) Cellular localization of interleukin 6 mRNA and interleukin 6 receptor mRNA in rat brain. Eur J Neurosci 5:1426–1435
Shalit F, Sredni B, Stern L et al (1994) Elevated interleukin-6 secretion levels by mononuclear cells of Alzheimer's patients. Neurosci Lett 174:130–132
Shapiro LS, Farrell J, Haghighi AB (2012) Tocilizumab treatment for neuro-Behcet's disease, the first report. Clin Neurol Neurosurg 114:297–298
Shaw PJ, Bates D, Cartlidge NE et al (1987) Long-term intellectual dysfunction following coronary artery bypass graft surgery: a six month follow-up study. Q J Med 62:259–268
Shibata N, Ohnuma T, Takahashi T et al (2002) Effect of IL-6 polymorphism on risk of Alzheimer disease: genotype-phenotype association study in Japanese cases. Am J Med Genet 114:436–439
Shrikant P, Weber E, Jilling T, Benveniste EN (1995) Intercellular adhesion molecule-1 gene expression by glial cells. Differential mechanisms of inhibition by IL-10 and IL-6. J Immunol 155:1489–1501
Silvestroni A, Faull RLM, Strand AD, Möller T (2009) Distinct neuroinflammatory profile in post-mortem human Huntington's disease. Neuroreport 20:1098–1103
Singh VK, Guthikonda P (1997) Circulating cytokines in Alzheimer's disease. J Psychiatr Res 31:657–660
Spronk PE, ter Borg EJ, Limburg PC, Kallenberg CG (1992) Plasma concentration of IL-6 in systemic lupus erythematosus; an indicator of disease activity? Clin Exp Immunol 90:106–110
Steffensen SC, Campbell IL, Henriksen SJ (1994) Site-specific hippocampal pathophysiology due to cerebral overexpression of interleukin-6 in transgenic mice. Brain Res 652:149–153
Stojanovic A, Martorell L, Montalvo I et al (2014) Increased serum interleukin-6 levels in early stages of psychosis: associations with at-risk mental states and the severity of psychotic symptoms. Psychoneuroendocrinology 41:23–32
Strauss S, Bauer J, Ganter U et al (1992) Detection of interleukin-6 and alpha 2-macroglobulin immunoreactivity in cortex and hippocampus of Alzheimer's disease patients. Lab Invest 66:223–230
Stys PK, Jiang Q (2002) Calpain-dependent neurofilament breakdown in anoxic and ischemic rat central axons. Neurosci Lett 328:150–154
Suzuki S, Tanaka K, Nagata E et al (1999) Cerebral neurons express interleukin-6 after transient forebrain ischemia in gerbils. Neurosci Lett 262:117–120
Tancredi V, D'Antuono M, Cafè C et al (2000) The inhibitory effects of interleukin-6 on synaptic plasticity in the rat hippocampus are associated with an inhibition of mitogen-activated protein kinase ERK. J Neurochem 75:634–643
Tarkowski E, Blennow K, Wallin A, Tarkowski A (1999) Intracerebral production of tumor necrosis factor-alpha, a local neuroprotective agent, in Alzheimer disease and vascular dementia. J Clin Immunol 19:223–230
Tarkowski E, Liljeroth AM, Minthon L et al (2003) Cerebral pattern of pro- and anti-inflammatory cytokines in dementias. Brain Res Bull 61:255–260
Tatemichi TK, Desmond DW, Stern Y et al (1994) Cognitive impairment after stroke: frequency, patterns, and relationship to functional abilities. J Neurol Neurosurg Psychiatr 57:202–207
Toulmond S, Vige X, Fage D, Benavides J (1992) Local infusion of interleukin-6 attenuates the neurotoxic effects of NMDA on rat striatal cholinergic neurons. Neurosci Lett 144:49–52
Trysberg E, Carlsten H, Tarkowski A (2000) Intrathecal cytokines in systemic lupus erythematosus with central nervous system involvement. Lupus 9:498–503
Urbaniak P, Hasler P, Kretzschmar S (2012) Refractory neuro-Behçet treated by tocilizumab: a case report. Clin Exp Rheumatol 30:S73–5
Vallières L, Campbell IL, Gage FH, Sawchenko PE (2002) Reduced hippocampal neurogenesis in adult transgenic mice with chronic astrocytic production of interleukin-6. J Neurosci 22:486–492
Vallières L, Rivest S (1997) Regulation of the genes encoding interleukin-6, its receptor, and gp130 in the rat brain in response to the immune activator lipopolysaccharide and the proinflammatory cytokine interleukin-1beta. J Neurochem 69:1668–1683
van Duijn CM, Hofman A, Nagelkerken L (1990) Serum levels of interleukin-6 are not elevated in patients with Alzheimer's disease. Neurosci Lett 108:350–354
Varghese JN, Moritz RL, Lou M-Z et al (2002) Structure of the extracellular domains of the human interleukin-6 receptor alpha -chain. Proc Natl Acad Sci USA 99:15959–15964
Visser M, Pahor M, Taaffe DR et al (2002) Relationship of interleukin-6 and tumor necrosis factor-alpha with muscle mass and muscle strength in elderly men and women: the Health ABC Study. J Gerontol A Biol Sci Med Sci 57:M326–32
Wada-Isoe K, Wakutani Y, Urakami K, Nakashima K (2004) Elevated interleukin-6 levels in cerebrospinal fluid of vascular dementia patients. Acta Neurol Scand 110:124–127
Wallenius K, Wallenius V, Sunter D et al (2002) Intracerebroventricular interleukin-6 treatment decreases body fat in rats. Biochem Biophys Res Commun 293:560–565
Wang J, Dunn AJ (1998) Mouse interleukin-6 stimulates the HPA axis and increases brain tryptophan and serotonin metabolism. Neurochemistry International 33:143–154
Waterloo K, Omdal R, Husby G, Mellgren SI (2002) Neuropsychological function in systemic lupus erythematosus: a five-year longitudinal study. Rheumatology (Oxford) 41:411–415
Weaver JD, Huang M-H, Albert M et al (2002) Interleukin-6 and risk of cognitive decline: MacArthur studies of successful aging. Neurology 59:371–378
Wei H, Alberts I, Li X (2013) Brain IL-6 and autism. Neuroscience 252:320–325
Wood JA, Wood PL, Ryan R et al (1993) Cytokine indices in Alzheimer's temporal cortex: no changes in mature IL-1 beta or IL-1RA but increases in the associated acute phase proteins IL-6, alpha 2-macroglobulin and C-reactive protein. Brain Res 629:245–252
Wu Y, Shaghaghi EK, Jacquot C et al (1999) Synergism between interleukin-6 and interleukin-1beta in hypothalamic serotonin release: a reverse in vivo microdialysis study in F344 rats. Eur Cytokine Netw 10:57–64
Xiao E, Xia L, Ferin M, Wardlaw SL (1999) Intracerebroventricular injection of interleukin-1 stimulates the release of high levels of interleukin-6 and interleukin-1 receptor antagonist into peripheral blood in the primate. Journal of Neuroimmunology 97:70–76
Yaffe K, Kanaya A, Lindquist K et al (2004) The metabolic syndrome, inflammation, and risk of cognitive decline. JAMA 292:2237–2242
Yamada M, Hatanaka H (1994) Interleukin-6 protects cultured rat hippocampal neurons against glutamate-induced cell death. Brain Res 643:173–180
Yamakawa Y, Sugita Y, Nagatani T et al (1996) Interleukin-6 (IL-6) in patients with Behçet's disease. J Dermatol Sci 11:189–195
Zalcman S, Green-Johnson JM, Murray L et al (1994) Cytokine-specific central monoamine alterations induced by interleukin-1, −2 and −6. Brain Res 643:40–49
Zhang J, Terreni L, De Simoni MG, Dunn AJ (2001) Peripheral interleukin-6 administration increases extracellular concentrations of serotonin and the evoked release of serotonin in the rat striatum. Neurochemistry International 38:303–308
Zhao S-J, Guo C-N, Wang M-Q et al (2012) Serum levels of inflammation factors and cognitive performance in amnestic mild cognitive impairment: a Chinese clinical study. Cytokine 57:221–225
Zhong J, Dietzel ID, Wahle P et al (1999) Sensory impairments and delayed regeneration of sensory axons in interleukin-6-deficient mice. J Neurosci 19:4305–4313
Zuliani G, Guerra G, Ranzini M et al (2007a) High interleukin-6 plasma levels are associated with functional impairment in older patients with vascular dementia. Int J Geriatr Psychiatry 22:305–311
Zuliani G, Ranzini M, Guerra G, et al. (2007b) Plasma cytokines profile in older subjects with late onset Alzheimer’s disease or vascular dementia. J Psychiatr Res 41:686–693
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Trapero, I., Cauli, O. Interleukin 6 and cognitive dysfunction. Metab Brain Dis 29, 593–608 (2014). https://doi.org/10.1007/s11011-014-9551-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-014-9551-2