
Abstract
Exposure to 3, 4-methylenedioxymethamphetamine (MDMA) leads to spatial memory impairment and hippocampal cell death. In the present study we have examined the protective effects of N-acetyl-L-cysteine (NAC) on MDMA-induced neurotoxicity. A total of 56 male Sprague Dawley rats (200–250 g) received twice daily intraperitoneal (IP) injections of 5, 10 or 20 mg/kg MDMA plus NAC (100 mg/kg). Rectal temperatures were recorded before and after daily treatment. We used a Morris water maze (MWM) to assess spatial learning and memory. At the end of the study rats’ brains were removed, cells were counted and the level of Bcl-2, Bax and caspase-3 expression in the hippocampi were measured. NAC pretreatment significantly reduced MDMA-induced hyperthermia. In the MWM, NAC significantly attenuated the MDMA-induced increase in distance traveled; however the observed increase in escape latency was not significant. The decrease in time spent in the target quadrant in MDMA animals was significantly attenuated (p < 0.001, all groups). NAC protected against MDMA-induced cell death and the up -regulation of Bax and Caspase-3, in addition to the down-regulation of Bcl-2. This data suggested a possible benefit of NAC in the treatment of neurotoxicity among those who use MDMA.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
3, 4-methylendioxymethamphetamine (MDMA) or ecstasy, is an amphetamine analog used as a recreational drug. Generally, MDMA causes elevated mood and a heightened sense of empathy (Fcology Arre et al. 2004). Ecstasy is capable of producing both reversible and reversible brain changes, such as expression of microglia cells and the hypertrophy of astrocytes, and neuronal degeneration in the various areas of the brain(For review see Sarkar and Schmued 2010). It has been reported that degenerating pyramidal and non pyramidal neurons were localized within the parietal cortex, tenia tecta, and thalamic nuclei (Schmued 2003). MDMA can also result in degenerating neurons in the insular and perirhinal cortex (Bowyer et al. 2008). MDMA has excitatory effects on the central nervous system, mostly in the serotonergic, dopaminergic and noradrenergic synaptic endings. The highest affinity is noted for the serotonin transporter (SERT) and 5-hydroxytryptamine 2 (5-HT2) receptors (Simantov and Tauber 1997). The neurotoxic effect of MDMA has been reported to be caused by acute release of 5-hydroxytryptamine (5-HT) from nerve endings, as well as destruction of 5-HT axons and 5-HT transporters involved in the development of hyperthermic responses and hyperactivity (O'Leary et al. 2001). Previous experimental studies have shown that MDMA-induced neurotoxicity is characterized by functional impairment in memory and apoptosis (Kuypers et al. 2011 and Soleimani Asl et al. 2012). It has been shown that hydroxyl radicals and lipid peroxidation following MDMA treatment induce serotonergic neurotoxicity (Sprague and Nichols 1995). Oxidative stress and hyperthermic responses following MDMA treatment involve MDMA-induced neurotoxicity that lead to the formation of hydroxyl radicals, lipid peroxidation and cell death (Shankaran et al. 1999; Alves et al. 2009). Oxidative stress is one important mechanism responsible for the disruption of the blood brain barrier (BBB) that leads to passage of toxic substances into the brain, ultimately resulting in the development and progression of various neurological diseases (Lee et al. 2012).
Oxidative stress is generated by an imbalance between reactive oxygen species (ROS) and antioxidants. Glutathione (GSH) is an important intracellular antioxidant that protects cells against oxidative stress (Franco and Cidlowski 2012).
As glutathione is unable to cross the BBB, possible treatment options would include the glutathione analog as a suitable candidate for therapeutic applications (Slivka et al. 1987). A thiol-based antioxidant has shown efficacy in the treatment of various neurological disorders. N-acetyl-L-cysteine (NAC), a novel thiol antioxidant, is a glutathione precursor that has the capability to freely cross the BBB (Cuzzocrea et al. 2000). NAC is used clinically for detoxification following acetaminophen poisoning and adult respiratory distress syndrome. The reducing thiol group in NAC reacts with hydroxyl radicals, scavenges free radicals, and increases both intracellular cysteine and cellular antioxidant levels (De Flora et al. 2001). Studies show that NAC can improve memory deficits of learning in mice (Zhang et al. 1994) and decrease the apoptotic response following radiotherapy (Xue et al. 2011). Neuroprotective effects of NAC were observed in different neuronal and glial in vitro models and glial cells against combined exposure to simulated ischemia and arachidonic acid (Gabryel et al. 2005). Studies using the rat model have shown that treatment with NAC to be protective against methamphetamine-induced hyperactivity, in addition to exerting protective effects against neurotoxicity (Fukami et al. 2004).
In the present study, we seek to determine if NAC can improve MDMA-induced neurotoxicity in male rats.
Materials and methods
Materials
We obtained NAC from Sigma Company (Seelze, Germany); MDMA was procured from the Presidency Drug Control Headquarters (Tehran, Iran). Other chemicals were purchased from commercial sources.
Animals and experimental protocol
We included 56 male Sprague Dawley rats, weighing 200–250 g (Razi Institute, Iran), in this experimental study. The rats were allowed to acclimatize to the colony room for 1 week prior to administration of MDMA. As MDMA causes hyperthermia, any animal that exhibited a high body temperature was excluded. Rats were maintained in the colony room at a temperature of 21 ± 1 °C, and the relative humidity of 50 ± 5 % on a 12-h light/12-h dark cycle with access to water and food ad libitum. All experimental procedures were performed in accordance with the Guidelines of the ethical Committee of Tehran University of Medical Sciences.
Animals were assigned to the following groups:
-
1.
Sham saline group (n = 7) received intraperitoneal (IP) injections of 1 mls normal saline, twice daily for 1 week.
-
2.
NAC control group (n = 7) received intraperitoneal (IP) injections of 100 mg/kg NAC, twice daily for 1 week.
-
3.
The experimental group was subdivided into three groups (n = 7 per group). Each group received MDMA at doses of 5, 10 or 20 mg/kg (IP), twice daily for 1 week (Soleimani Asl et al. 2011).
-
4.
The treatment group was subdivided into three groups (n = 7 per group). Each group received IP administrations of 100 mg/kg NAC 1 h prior to administration of MDMA (5, 10, or 20 mg/kg), twice daily for 1 week.
Body temperature monitoring
Rectal temperature of all animals was daily monitored 30 min before NAC treatment and 1 h after MDMA treatment by digital thermometer over the treatment period (Flex Temp ІІ, Omeron, Veghel, and Netherlands) following the protocol outlined by the manufacturer.
Learning memory was assessed by the Morris water maze (MWM) on the same day following the last drug administration.
Morris water maze (MWM)
We used this apparatus to assess spatial memory. The MWM consisted of a circular pool (180 cm in diameter, 60 cm in height) that was painted black and filled to a depth of 35 cm with water at a temperature of 22 ± 1 °C. The pool was divided into four quadrants with four starting locations, which were referred to as north (N), east (E), south (S) and west (W), each located at equal distances along the pool rim.
An invisible platform (10 cm diameter) constructed from Plexiglass was located 1 cm below the water in the center of the northern quadrant. We trained the animals for 3 days at approximately the same time (10:00–12:00 a.m.) each day. Each training day included two blocks, with four trials. The time limit on each trial was 90 s. There was a 30-s period between two trials, which was spent on the platform. Rats were allowed to rest for 5 min between two consecutive blocks. A video camera (Nikon, Melville, NY, USA) linked to a computer was mounted directly above the water maze pool to record the time taken to reach the hidden platform (escape latency), the length of the swim path (traveled distance) and percentage of time spent in the target quadrant for each rat. The day after the last learning trial, each rat was given a single 60-s probe trial and a visible test. The probe trials were conducted without a platform. In the visible trials, the platform was covered with aluminum foil.
Histology
For the light microscopy study, we prepared the sections using our previously described method (Mehdizadeh et al. 2012). In brief, rats were perfused with 4 % paraformaldehyde in 0.1 M phosphate buffer (pH 7.3) and their hippocampi were serially sectioned into 10 μm coronal sections. After deparaffinization and rehydration, sections were stained in 0.1 % cresyl violet for 3 min. Finally, the sections were photographed with a digital camera (Olympus, DP 11, Japan) attached to a microscope (Olympus Provis, Ax70, Japan). For each animal, the average neuronal counts were obtained by counting five serial coronal sections at 120 μm intervals at 400× magnification. An experienced histologist unaware of the treatment conditions performed the histological assessments. Only intact neuronal cells with clearly defined cell bodies and nuclei were counted.
Western blot
After the MWM, the animals were killed by cervical dislocation. Animals’ brains were rapidly removed and the hippocampi dissected out on ice, then frozen in liquid nitrogen and maintained at −80 °C until used. The frozen hippocampi were homogenized with ice-cold lysis buffer that contained Ripa buffer with a protease inhibitor cocktail (1:10) for 1 h and centrifuged (Eppendorf, Hamburg, Germany) at 12,000 g for 20 min at a temperature of 4 °C, after which the supernatant was removed and stored. After determining the protein concentration with a Bio-Rad assay system (Bio-Rad, San Francisco, CA, USA), 100 μg protein aliquots from each sample were denatured with sample buffer (6.205 mM Tris–HCl, 10 % glycerol, 2 % SDS, 0.01 % bromophenol blue and 50 mM 2-ME) at 95 °C for 5 min, then separated on a 10 % sodium dodecyl sulphate polyacrylamide gel for 90 min at 120 V. Next, proteins were transferred to a Hybond-P™ membrane (Amersham Pharmacia Biotech, Piscataway, NJ, USA). Membranes were blocked with 5 % nonfat milk dissolved in TTBS buffer that consisted of tris (50 mM), 1.5 % NaCl and 0.05 % Tween 20 at pH 7.5 for 1 h. Membranes were incubated with monoclonal antibody to Bcl-2 and Bax, and polyclonal antibody to caspase-3 (1:1000; Sigma Aldrich, St, Louis, MO, USA) for 2 h and alkaline phosphatase-conjugated secondary antibody (1:10000; Sigma Aldrich, St. Louis, MO, USA) for 1 h. Bands were detected by incubation with chromogenic substrate 5-bromo-4-chloro-3-indolyl phosphate in the presence of nitroblue tetrazolium. Β-actin antibody (1:1000; Sigma Aldrich, St. Louis, MO, USA) was used to detect endogenous standard for normalization. The bands from various groups that corresponded to the appropriate molecular weight for each subunit were analyzed and values compared using densitometric measurements by an image analysis system (UVIdoc, Houston, TX, USA).
Reverse transcriptase PCR
We used phenol-chloroform extract to remove total mRNA from the frozen hippocampi. Tissue samples were homogenized in 1,000 μl RNA™ (Cinnagen, Tehran, Iran) after which 200 μl of ice-cold chloroform was added. The homogenates were centrifuged (Eppendorf, Hamburg, Germany) at 12,000 g for 20 min at 4 °C. The RNA of the water-soluble supernatant was precipitated with isopropanol and washed with 75 % ethanol. The air-dried RNA pellet was dissolved in RNase-free water. cDNA first-strand synthesis was performed by a cDNA synthesis Kit (Quiagen, Hilden, Germany) following the protocol outlined by the manufacturer. First strand cDNA (2.5 ml) was used as a template for subsequent PCR with a PCR Master Kit (Cinnagen, Tehran, Iran) and the following primers (Cinnagen, Tehran, Iran): β-actin forward (5′TGGAGAAGAGCTATGAGCTGCCTG′3) and reverse (5′GTGCCACCAGACAGCACTGTGTTG′3); Bax forward (5′CCAAGAAGCTGAGCGAGTGTCTC′3) and reverse (5′AGTTGCCATCAGCAAACATGTCA′3); Bcl-2 forward (5′CGCCCGCTGTGCACCGAGA ′3) and reverse (5′CACAATCCTCCCCCAGTTCACC′3); and caspase3 forward (5TTTGGAACGAACGGACCTGT3) and (reverse 5CACGGGATCTGTTTCTTTGC3).
For PCR, we prepared a 25 μl reaction mixture that contained 1 μl cDNA, 12.5 μl Master Mix, 9.5 μl sterile deionized water and 1 μl each of the forward and reverse primers. The PCR reactions were programmed as follows: For Bax was used initial denaturation at 95 °C for 3 min, followed by 31 cycles at 95 °C for 20 s, 65 °C for 30 s and 72 °C for 30 s, and finally elongation time at 72 °C for 7 min. For Bcl-2, we used initial denaturation at 95 °C for 3 min, 35 cycles at 95 °C for 30 s, 60 °C for 1 min and 72 °C for 60 min. Initial denaturation at 95 °C for 3 min, 35 cycles at 95 °C for 50 s, 58 °C for 45 s and 72 °C for 50 s were used for caspase3, and the reactions were terminated by elongation period at 72 °C for 7 min. The same annealing temperature was used for β-actin. PCR products were separated by electrophoresis in 1.5 % agarose gel at 100 V. Semi-quantitative analysis was assessed by a digital imaging system (UVIdoc, Houston, TX, USA).
Statistical analysis
Data were presented as means ± S.E.M and results analyzed by SPSS 16 software. One-way ANOVA and post-hoc comparison were performed using the Tukey and two-way repeated measurements tests. P ≤ 0.05 was considered statistically significant.
Results
Body temperature
NAC attenuated MDMA-induced hyperthermia. Prior to the injections, both MDMA and the sham groups had initial temperatures of 36–37 °C. Analysis of pre- and post-treatment temperature data of repeated measurements showed increased rectal temperatures in all MDMA groups compared with the sham group (Fig. 1, P < 0.001). These increases were dose-dependent. Injection of NAC 60 min prior to MDMA administration inhibited the increase in body temperature caused by MDMA [Fig. 1, P < 0.01 for NAC + MDMA (5 and10 mg/kg) and P < 0.05 for NAC + MDMA (20 mg/kg)].

Protective effect of NAC on MDMA-induced hyperthermia. MDMA caused significant increase in body temperature 1 h after drug administration (*P < 0.001 vs. sham), which was dose-dependent. @P < 0.01 vs. MDMA (10 and 20 mg/kg). NAC pretreatment caused decrease in body temperature compared with the MDMA groups [•P < 0.001 vs. MDMA (5 and 10 mg/kg); #P < 0.01 vs. MDMA (20 mg/kg)]
Learning memory
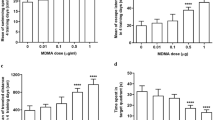
Results from the MWM showed that NAC improved MDMA-induced learning memory impairment.
According to the results, dose-dependent administration of MDMA increased escape latency (P < 0.001, all groups) and traveled distance (P < 0.01, all groups) during the training days (Fig. 2). NAC pretreatment caused insignificant reductions in escape latency and significant reductions in traveled distance compared with the MDMA groups (P < 0.01, all groups).

Protective effect of NAC on MDMA-induced learning and memory impairment in the MWM. MDMA caused an increase in escape latency (*P < 0.001 vs. sham group) and traveled distance (*P < 0.01 vs. sham group). MDMA caused a reduction in time spent in target quadrant (*P < 0.001 vs. sham group). NAC administration improved the mentioned parameters, which was significant for traveled distance (•P < 0.01 vs. MDMA groups)
There were no significant differences in escape latency and traveled distance in the visible trial session between the treatment and sham groups (data not shown).
There was a significant difference in time spent in the target quarter (quarter where the platform was located during the training days) in the probe trial session between the MDMA groups and sham group (Fig. 2, P < 0.001, all groups). Pretreatment with NAC attenuated the effect of MDMA to decrease the time spent in the target area, however it was not significant.
Histological analysis
Figure 3a–h shows coronal sections of cells in the CA1 hippocampus. Light microscopy of the sections stained with Nissl stain showed that MDMA significantly reduced neural density in the CA1 hippocampus compared to the sham group (Fig. 4, P < 0.001, all groups). Administration of NAC 60 min before MDMA treatment significantly decreased cell loss caused by MDMA [P < 0.05 for MDMA (5 and 10 mg/kg), P < 0.001 for MDMA (20 mg/kg)].

Nissl staining in sham (a), MDMA 5 mg/kg (b), MDMA 10 mg/kg (c), MDMA 20 mg/kg (d), MDMA (5 mg/kg) plus NAC (100 mg/kg) (e), MDMA (10 mg/kg) plus NAC (100 mg/kg) (f), MDMA (20 mg/kg) plus NAC (100 mg/kg) (g) groups, and NAC(100 mg/kg). After MWM, brains were fixed and serially sectioned into 10 μm coronal sections and stained in 0.1 % crysel violet. Magnification: 400×. Bar = 50 μm. White arrow shows intact cells. Black arrows represent dead cells

Protective effect of NAC on MDMA-induced cell death in CA1 hippocampus. MDMA treatment caused to a reduction in cell density (*P < 0.001 vs. sham group) and NAC pretreatment led to an increase in cell density compared with MDMA groups [#P < 0.05 vs. MDMA (5 and 10 mg/kg), @ P < 0.001 vs. MDMA (20 mg/kg)]
Analysis of Bax and Bcl-2 expression
Different doses of MDMA caused down-regulation of the Bcl-2 protein, which was only significant for the 20 mg/kg MDMA group compared with the sham group (P < 0.05). Pretreatment with NAC increased Bcl-2 protein expression compared to the MDMA groups, but this increase was not significant (Fig. 5a).

Western blot (a) and RT-PCR (b) analysis of Bax and Bcl-2 expression in sham and treatment groups. a A significant difference in Bcl-2 expression between MDMA groups and sham group was noted (@ P < 0.05 vs. sham). Bax expression increased in the MDMA groups compared to the sham group (*P < 0.01 vs. sham). Administration of NAC up- and down- regulated Bcl-2 and Bax expression, respectively. b MDMA caused a decrease in bcl-2 gene expression (*P < 0.05 vs. sham). MDMA treatment led to increased Bax expression (®P < 0.01 and #P < 0.001 vs. sham group). NAC pretreatment caused to an increase in bcl-2 and a decrease in bax expression compared with MDMA groups
As seen in Fig. 5a, there was a significant increase in Bax expression in the MDMA groups compared to the sham group (P < 0.01, all groups). NAC pretreatment decreased Bax expression compared with the MDMA groups, but this decrease was not significant.
Densitometry from the electrophoresis gel showed significantly more expression of the Bcl-2 gene in the sham group than the other groups (P < 0.05, all groups, Fig. 5b). NAC pretreatment caused increased Bcl-2 expression compared to the MDMA groups, but this increase was not significant.
There was a significant difference in Bax expression between the sham and the MDMA 10 and 20 mg/kg groups (P < 0.001) as well as the 5 mg/kg group (P < 0.01, Fig. 5b).
Administration of NAC 60 min before MDMA resulted in an insignificant decrease in Bax expression compared to the MDMA groups.
Bcl-2/Bax ratios
Cell survival depends on a molecular balancing act that regulates the ratio of Bcl-2 to Bax (Oltvai et al. 1993). Because apoptosis results from changing in this ratio, we opted to generate relative ratio based on the pattern of protein expression obtained from the western blot and RT-PCR. Figure 6a shows there were a significant reduction in the Bcl-2/Bax proteins ratio in MDMA groups compared with sham group (p < 0.001 for all groups). NAC treatment caused to an increase in this ratio when compared with MDMA- treated rats that was only significant in MDMA5mg/kg group (p < 0.01). As shown in Fig. 6b, there were significant differences in the bcl-2/bax genes ratio between sham and other groups (p < 0.001). NAC treatment caused to an insignificant increase in this ratio compared with MDMA groups.

Ratio of Bcl-2 to Bax protein (a) and gene (b) levels. The pattern suggest that MDMA injections led to a reduction in bcl-2/bax ratio (*P < 0.001 vs. sham) and NAC pretreatment increased this ratio (@ p < 0.01 vs. MDMA 5 mg/kg)
Analysis of caspase-3 expression
Caspase- 3 is one of the key executioners of apoptosis that synthesized as inactive proforms (procaspases) which upon receiving an apoptotic signal is cleaved and forms the active enzyme.
Different doses of MDMA significantly caused to up-regulation of the caspase 3 compared with the sham group (P < 0.001, all groups, Fig. 7a). Furthermore, there was significant differences between MDMA 5 and 10 mg/kg groups and MDMA 20 mg/kg group (p < 0.01). As seen in Fig. 7a, pretreatment with NAC significantly decreased caspase 3 protein expression compared with the MDMA groups (p < 0.001 for MDMA 5 and 20 mg/kg, p < 0.01 for MDMA 10 mg/kg).

Western blot (a) and RT-PCR (b) analysis of caspase 3 expression in sham and treatment groups. a A significant difference in caspase expression between sham group and others group was noted (* P < 0.001 vs. sham). Caspase 3 expressed more in MDMA 20 mg/kg compared with MDMA5 and 10 mg/kg (@ p < 0.01 vs. MDMA 20 mg/kg). Administration of NAC down- regulated caspase 3 expression (# p < 0.001 vs. MDMA 5 mg/kg, $ p < 0.01 vs. MDMA 10 mg/kg, &p < 0.001 vs. MDMA 20 mg/kg). b MDMA caused an increase in caspase 3 gene expression (*P < 0.001 vs. sham). There was significant differences between MDMA- treated groups (@ p < 0.01 vs. MDMA 20 mg/kg). NAC pretreatment led to decrease in caspase 3 expression (# p < 0.001 vs. MDMA 5 mg/kg, $ p < 0.01 vs. MDMA 10 mg/kg, &p < 0.001 vs. MDMA 20 mg/kg)
Densitometry from the electrophoresis gel showed significantly less expression of the caspase 3 gene in the sham group than the other groups (P < 0.001, all groups, Fig. 7b). Administration of NAC 60 min before MDMA caused to significant reduction in caspase 3 expression compared to the MDMA groups (p < 0.001 for MDMA 5 and 20 mg/kg, p < 0.01 for MDMA 10 mg/kg).
Discussion
The major findings of this study were the attenuation of memory impairment and hyperthermia by NAC following MDMA treatment. Pretreatment with NAC was protective against MDMA- induced neurotoxicity in the hippocampus.
The brain is sensitive to oxidative stress because of low antioxidant and cell membrane lipids (Viegas et al. 2012). Oxidative stress results from an imbalance between ROS and intracellular antioxidants. Therefore, the use of an external antioxidant is one of the most common therapeutic strategies for the treatment of neurotoxicity. NAC is a small molecule that contains a thiol group, which is used as an antioxidant. It scavenges free radicals and directly reacts with hydroxyl radicals (Cuzzocrea et al. 2000).
Several lines of evidence suggest that oxidative stress and hyperthermia play a role in MDMA- induced neurotoxicity in the brain. Previous studies suggest that MDMA causes temperature dysregulation which disturbs homeostasis and prevents an animal from maintaining a constant body temperature (Navarrete et al. 2007; Sprague et al. 2003). The serotonergic system plays an important role in body temperature homeostasis. Since MDMA causes to neurotoxicity of serotonergic system in the brain; possibly serotonin is responsible for MDMA-induced hyperthermia. MDMA causes vasoconstriction of peripheral vessels, inhibition of body temperature dissemination, and heat production via the mitochondrial activity of UCP3 (Pedersen and Blessing 2001; Mills et al. 2004).
Our results showed that NAC could decrease MDMA-induced hyperthermia which was consistent with results from the study by Fukami et al. (2004). Possibly this occurred by scavenging peripheral vasoconstriction, which enhanced mitochondrial activity and free radical scavenging.
NAC acts as a GSH precursor that improves beta amyloid peptide-induced learning and memory deficits in mice (Fu et al. 2006). This study confirmed our results in which NAC, as a glutathione precursor improved MDMA-induced memory impairments. NAC pretreatment caused an insignificant reduction in escape latency and a significant reduction in traveled distance compared with MDMA groups. It has been shown that in the MWM, escape latency and travelled distance are highly correlated (Vorhees et al. 2004),but our results showed non-significant and significant reductions in escape latency and traveled distance following NAC treatment, respectively; it may be an effect of NAC on swim speed.
The protective effects of NAC on memory are related to its function of scavenging free radicals. NAC is deacetylated to l-cysteine inside the cells and therefore enhances the rate of GSH, and subsequently potentiating the natural antioxidant cellular defense mechanisms (Cuzzocrea et al. 2000). Glutathione has excitatory effects on serotonergic endings and serotonin has a modulatory effect on long term potentiation (LTP) in the hippocampus, an important structure involved in memory (Slivka et al. 1987). In addition to the serotonergic system, glutathione plays a role in the process of cognition and memory (Pinnen et al. 2011; Choy et al. 2010).
As with the spatial memory results, our histological evaluation has shown that MDMA reduced the numbers of cells in the CA1 hippocampus. Administration of NAC attenuated cell death in the CA1 hippocampus which confirmed the results of other studies (Fu et al. 2006). Lan et al. have reported that NAC could prevent apoptotic death of neuronal cells and promote survival of pcl2 cells (Lan et al. 2011).
Cell death can be classified into necrosis and apoptosis or programmed cell death (Kroemer et al. 2009). Caspases are a family of cysteine proteases that play essential roles in programmed cell death (apoptosis). There are two types of apoptotic caspases: initiator caspases and executioner caspases. Initiator caspases cleave inactive pro-forms of executioner caspases, thereby activating them. Executioner caspases in turn cleave other protein substrates within the cell, to trigger the apoptotic process (for review see Miura 2012). Caspase 3 as an executioner caspases involves in the execution of apoptosis. Consistent with Jimenez et al. study (Jiménez et al. 2004), our results showed that MDMA treatment caused to up-regulation of caspase-3 expression, so it shows that MDMA-induced cell death has apoptosis Characteristic (Kermanian et al. 2012).
Studies showed up-regulation of the Bax protein and down-regulation of the Bcl-2 protein following treatment with amphetamine-derivatives (Jayanthi et al. 2001; Soleimani Asl et al. 2012). Montgomery et al. have shown that MDMA causes rapid intracellular Ca2+ influx, mitochondrial membrane depolarization, ROS production and caspase 9 activation (Montgomery et al. 2010). It seems the mechanism of neurotoxicity involves formation of metabolic products such as N-methyl-a-methyl dopamine (MeDA), orthoquinones and quinine thioethers (Capela et al. 2007). MDMA is converted to MeDA via isoenzyme cytochrome p-450 catabolism which is unstable and is metabolized in the presence of NADPH into quinine that reacts with glutathione, subsequently generating ROS which is a key factor of MDMA-induced neurotoxicity (Yamamoto and Raudensky 2008).
In addition, it has been reported that expression of the tumor necrosis factor (TNF) -α gene was induced after MDA administration and other amphetamine derivates (Gonçalves et al. 2008) which lead to decrease in intracellular glutathione. Pretreatment with NAC prevented a TNF-α-induced decrease in glutathione and streptozotocin-induced encephalopathy (Hashimoto et al. 2001; Kamboj et al. 2008). Taken together, it is possible that NAC inhibits inflammatory genes and ROS generation and might be implicated in the protection of amphetamine-induced neurotoxicity. In conclusion our findings have demonstrated that pretreatment with NAC significantly attenuated traveled distance reduction following MDMA treatment and improved MDMA- induced escape latency and time spent deficiencies, in addition to the hyperthermia associated with MDMA treatment. NAC has shown a protective effect against MDMA-induced neurotoxicity in the rat hippocampus. Therefore, it is likely that NAC may be useful as a potential treatment for the adverse effects associated with MDMA.
References
Alves E, Summavielle T, Alves CJ, Custódio J, Fernandes E, De Lourdes Bastos M (2009) Ecstasy-induced oxidative stress to adolescent rat brain mitochondria in vivo: influence of monoamine oxidase type. An Addict Biol 14:185–193
Bowyer JF, Thomas M, Schmued LC, Ali SF (2008) Brain region-specific neurodegenerative profiles showing the relative importance of amphetamine dose, hyperthermia, seizures, and the blood–brain barrier. Ann N Y Acad Sci 1139:127–139
Capela JP, Macedo C, Branco PS, Ferreira LM, Lobo AM et al (2007) Neurotoxicity mechanisms of thioether ecstasy metabolites. Neuroscience 8:1743–1757
Choy KH, Dean O, Berk M, Bush AI, Van Den Buuse M (2010) Effects of N-acetyl-cysteine treatment on glutathione depletion and a short-term spatial memory deficit in 2-cyclohexene-1-one-treated rats. Eur J Pharmacol 15:224–228
Cuzzocrea S, Mazzon E, Costantino G, Serraino I, Dugo L, Calabrò G et al (2000) Beneficial effects of n-acetylcysteine on ischaemic brain injury. Br J Pharmacol 130:1219–1226
De Flora S, Izzotti A, D'Agostini F, Balansky RM (2001) Mechanisms of N-acetylcysteine in the prevention of DNA damage and cancer, with special reference to smoking-related end-points. Carcinogenesis 22:999–1013
Fcology Arre M, Mathuna BO, Roset PN et al (2004) Repeated doses administration of MDMA in humans: pharmacological effects and pharmacokinetics. Psychopharmacology (Berlin) 173(3–4):364–375
Franco R, Cidlowski JA (2012) Glutathione efflux and cell death. Antioxid Redox Signal 15:1694–1713
Fu AL, Dong ZH, Sun MJ (2006) Protective effect of N-acetyl-L-cysteine on amyloid beta-peptide-induced learning and memory deficits in mice. Brain Res 13:201–206
Fukami G, Hashimoto K, Koike K, Okamura N, Shimizu E, Iyo M (2004) Effect of antioxidant N-acetyl-L-cysteine on behavioral changes and neurotoxicity in rats after administration of methamphetamine. Brain Res 30:90–95
Gabryel B, Toborek T, Małecki A (2005) Immunosuppressive immunophilin ligands attenuate damage in cultured rat astrocytes depleted of glutathione and exposed to simulated ischemia in vitro. Comparison with N-acetylcysteine. Neurotoxicology 26(3):373–384
Gonçalves J, Martins T, Ferreira R, Milhazes N, Borges F, Ribeiro CF et al (2008) Methamphetamine-induced early increase of IL-6 and TNF-alpha mRNA expression in the mouse brain. Ann N Y Acad Sci 1139:103–111
Hashimoto S, Gon Y, Matsumoto K, Takeshita I, Horie T (2001) N-acetylcysteine attenuates TNF-alpha-induced p38 MAP kinase activation and p38 MAP kinase-mediated IL-8 production by human pulmonary vascular endothelial cells. Br J Pharmacol 132:270–276
Jayanthi S, Deng X, Bordelon M, McCoy MT, Cadet JL (2001) Methamphetamine causes differential regulation of pro-death and anti-death Bcl-2 genes in the mouse neocortex. FASEB J 15:1745–1752
Jiménez A, Jordà Elvira G, Verdaguer E, Pubill D, Sureda F, Canudas AM et al (2004) Neurotoxicity of amphetamine derivatives is mediated by caspase pathway activation in rat cerebellar granule cells. Follow Toxicol Appl Pharmacol 196:223–234
Kamboj SS, Chopra K, Sandhir R (2008) Neuroprotective effect of N-acetylcysteine in the development of diabetic encephalopathy in streptozotocin-induced diabetes. Metab Brain Dis 23(4):427–443
Kermanian F, Mehdizadeh M, Soleimani M, Ebrahimzadeh Bideskan AR et al (2012) The role of adenosine receptor agonist and antagonist on Hippocampal MDMA detrimental effects; a structural and behavioral study. Metab Brain Dis 27:459–469
Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH et al (2009) Classification of cell death: recommendations of the Nomenclature Committee on Cell Death Cell Death Differ. Cell Death Differ 16(1):3–11
Kuypers KP, Wingen M, Heinecke A, Formisano E, Ramaekers JG (2011) MDMA intoxication and verbal memory performance: a placebo-controlled pharmaco-MRI study. J Psychopharmacol 25(8):1053–1061
Lan A, Liao X, Mo L, Yang C, Yang Z, Wang X, Hu F et al (2011) Hydrogen sulfide protects against chemical hypoxia-induced injury by inhibiting ROS-activated ERK1/2 and p38MAPK signaling pathways in PC12 cells. PLoS One 6:e25921
Lee DH, Gold R, Linker RA (2012) Mechanisms of oxidative damage in multiple sclerosis and neurodegenerative diseases: therapeutic modulation via fumaric acid esters. Int J Mol Sci 13(9):11783–11803
Mehdizadeh M, Dabaghian F, Nejhadi A, Fallah-huseini H, Choopani S, Shekarriz N et al (2012) Zingiber officinale alters 3, 4-methylenedioxymetham-induced neurotoxicity in rat brain. Cell J 14(3):177–184
Mills EM, Rusyniak DE, Sprague JE (2004) The role of the sympathetic nervous system and uncoupling proteins in the thermogenesis induced by 3,4-methylenedioxymethamphetamine. J Mol Med (Berl) 82:787–799
Miura M (2012) Apoptotic and nonapoptotic caspase functions in animal development. Cold Spring Harb Perspect Biol 4(10):1
Montgomery T, Sitte H, McBean G (2010) 4-Methylthioamphetamine (4-MTA) induces mitochondrial-dependent apoptosis in SH-SY5Y cells independently of dopamine and noradrenaline transporters. BMC Pharmacology 10(supp 1):A22
Navarrete M, Núñez H, Ruiz S, Soto-Moyano R, Valladares L, White A et al (2007) Prenatal undernutrition decreases the sensitivity of the hypothalamo-pituitary-adrenal axis in rat, as revealed by subcutaneous and intra-paraventricular dexamethasone challenges. Neurosci Lett 29:99–103
O'Leary G, Nargiso J, Weiss RD (2001) 3, 4-methylenedioxymethamphetamine (MDMA): a review. Curr Psychiatr Rep 3(6):477–483
Oltvai ZN, Milliman CL, Korsmeyer SJ (1993) Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 74(4):609–619, 27
Pedersen NP, Blessing WW (2001) Cutaneous vasoconstriction contributes to hyperthermia induced by 3, 4-methylenedioxymethamphetamine (ecstasy) in conscious rabbits. J Neurosci 1:8648–8654
Pinnen F, Sozio P, Cacciatore I, Cornacchia C, Mollica A, Iannitelli A et al (2011) Ibuprofen and glutathione conjugate as a potential therapeutic agent for treating Alzheimer’s disease. Arch Pharm (Weinheim) 344:139–148
Sarkar S, Schmued L (2010) Neurotoxicity of ecstasy (MDMA): an overview. Curr Pharm Biotechnol 11(5):460–469
Schmued LC (2003) Demonstration and localization of neuronal degeneration in the rat forebrain following a single exposure to MDMA. Brain Res 974(1–2):127–133
Shankaran M, Yamamoto BK, Gudelsky GA (1999) Involvement of the serotonin transporter in the formation of hydroxyl radicals induced by 3, 4-methylenedioxymethamphetamine. Eur J Pharmacol 385:103–110
Simantov R, Tauber M (1997) The abused drug MDMA (Ecstasy) induces programmed death of human serotonergic cells. FASEB J 11(2):141–146
Slivka A, Mytilineou C, Cohen G (1987) Histochemical evaluation of glutathione in brain. Brain Res 21:275–284
Soleimani Asl S, Naghdi N, Choopani S, Farhadi MH, Samzadeh-Kermani A, Mehdizadeh M (2011) Non-acute effects of different doses of 3–4, methylenedioxymethamphetamine (MDMA) on spatial memory in the Morris water maze in Sprague Dawley male rats. NRR 6:1715–1719
Soleimani Asl S, Farhadi MH, Moosavizadeh K, Samadi Kuchak Saraei A, Soleimani M et al (2012) Evaluation of Bcl-2 family gene expression in hippocampus of 3–4 methylenedioxymethamphetamine treated rats. Cell J 13:275–280
Sprague JE, Nichols DE (1995) The monoamine oxidase-B inhibitor L-deprenyl protects against 3,4-methylenedioxymethamphetamine-induced lipid peroxidation and long-term serotonergic deficits. J Pharmacol Exp Ther 273(2):667–673
Sprague JE, Preston AS, Leifheit M, Woodside B (2003) Hippocampal serotonergic damage induced by MDMA (ecstasy): effects on spatial learning. Physiol Behav 79:281–287
Viegas CM, Tonin AM, Zanatta A, Seminotti B, Busanello EN, Fernandes CG et al (2012) Impairment of brain redox homeostasis caused by the major metabolites accumulating in hyperornithinemia-hyperammonemia-homocitrullinuria syndrome in vivo. Metab Brain Dis 27(4):521–530
Vorhees CV, Reed TM, Skelton MR, Williams MT (2004) Exposure to 3, 4-methylenedioxymethamphetamine (MDMA) on postnatal days 11–20 induces reference but not working memory deficits in the Morris water maze in rats: implications of prior learning. Int J Dev Neurosci 22:247–259
Xue C, Liu W, Wu J, Yang X, Xu H (2011) Chemoprotective effect of N-acetylcysteine (NAC) on cellular oxidative damages and apoptosis induced by nano titanium dioxide under UVA irradiation. Toxicol in Vitro 25:110–116
Yamamoto BK, Raudensky J (2008) The role of oxidative stress, metabolic compromise, and inflammation in neuronal injury produced by amphetamine-related drugs of abuse. J Neuroimmune Pharmacol 3:203–217
Zhang H, Spapen H, Nguyen DN, Benlabed M, Buurman WA, Vincent JL (1994) Protective effects of N-acetyl-L-cysteine in endotoxemia. Am J Physiol 266:1746–1754
Acknowledgments
This research was supported by a grant from Tehran University of Medical Sciences, No. P/664.
Conflict of interest statement
None of the authors of this paper have a financial interest to report.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Soleimani Asl, S., Mousavizedeh, K., Pourheydar, B. et al. Protective effects of N-acetylcysteine on 3, 4-methylenedioxymethamphetamine-induced neurotoxicity in male Sprague–Dawley rats. Metab Brain Dis 28, 677–686 (2013). https://doi.org/10.1007/s11011-013-9423-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-013-9423-1